









11. دوره کاربری دستگاه Real time PCR
آموزشگاه آزاد نانو زیست فناوری اوژن اولین آموزشگاه رسمی فنی و حرفه ای در رشته نانو زیست فناوری در تهران برگزار میکند:
کاربری دستگاه Real time PCR
سرفصل مطالب
- طراحی پرایمر
- تهیه و بهینه سازی مخلوط واکنش پلیمریزاسیون.
- برنامه دهی تکنیک pcr و روند پلیمریزاسیون
- تجزیه و تحلیل داده ها و آنالیز نمودار نتایج
- نگهداری و راه اندازی دستگاه Real time PCR
- کار با نرم افزارها و نرم افزارهای مرتبط مانند primer express, GeneRunner.
- به کارگیری ضوابط ایمنی و بهداشتی در محیط کار
- ظرفیت محدود
- همراه با ارائه مدرک معتبر
ویژگی های آموزشگاه نانو زیست فناوری اوژن (تکوین)
- تمام دوره ها و کارگاه ها با ظرفیت محدود (نهایتا ۴ تا ۵ نفر) برگزار می گردد.
- تمام تکنیک های عملی توسط خود کارآموز بعد از آموزش کامل انجام می شود.
- در پایان دوره های آموزشگاه اوژن می توانید در آزمون فنی حرفه ای شرکت کرده و مدرک فنی حرفه ای دریافت نمایید، که به علت قید ساعت آموزشی برای شما به منزله ی کارآموزی یا کارورزی بوده و در تمام دانشگاه ها دنیا معتبر و قابل ترجمه می باشد.
برای اطلاع از جزئیات ، ثبت نام و مشاوره می توانید از طریق شماره تلفن، واتساپ، اینستاگرام ، تلگرام و وبسایت آموزشگاه آزاد نانو زیست فناوری اوژن اقدام کنید.
www.ogene-tech.com
tel: 021-44961487-09120169816
whats app: 09233093463
t.me/Ogenetechnology
@ogenetech
مهارت شما آینده شماست
توضیحات
دوره کاربری دستگاه Real time PCR

اساس روش
real time PCR چیست؟ نوعی PCR می باشد که در آن توالی تکثیر شده به صورت کمی سنجیده می شود. برای این کار از دستگاه Real-time PCR(نوعی ترمال سایکلر) استفاده می شود که از یک سیستم اپتیکی بهره برده و میزان DNA هر نمونه توسط آن محاسبه می گردد.
در تکنیک Real Time PCR از یک مولکول گزارشگر فلوروسنت برای مشاهده پیشرفت PCR استفاده می شود و قدرت سیگنال فلوئورسنت تولید شده ارتباط مستقیمی با مقدار مولکول های تکثیرشده دارد. در تکنیک Real Time PCR میزان محصولاتی که حین یک آزمایش PCR تولید می شوند با میزان محصولات تولید شده طی آزمایش های PCR با مقدار نوکلئیک اسید آغازکننده مشخص، مقایسه میشود و امکان پی بردن به مقدار نوکلئیک اسید اولیه موجود در نمونه، برای ما فراهم می شود.
در وهله اول برای انجام آزمایش real-time PCR نیاز به نمونه هایی که سنتز cDNA شده اند داریم، بنابراین قبل از فرآیند سننتز cDNA بایستی از نمونه ها، RNA استخراج کرده باشیم و RNA ای استخراج شده را مورد سنجش غلظت یا خلوص قرار دهیم(اسپکتروفتومتری) که ببینیم نمونه های RNA که استخراج شدند قابلیت این که ما بخواهیم با آنها cDNA را سنتز کنیم، دارند یا خیر و در مرحله بعد سنتز cDNA را انجام می دهیم.
برای برای انجام آزمایش آنالیز بیان ژن باید mRNA را بررسی کنیم، چون mRNA ناپایدار است، و در مدت زمان کمی از بین می رود، به همین دلیل یکی از اهدافی که در فرآیند سنتز cDNA انجام می دهیم این است که mRNA را از حالت ناپایدار تبدیل به فرم پایدار کنیم تا مدت زمان نگهداری mRNA بیشتر شود.زمانی که سنتز cDNA انجام شد، پس از آماده سازی نمونه به منظور ورود به سیکل های پلیمریزه شدن، به نمونه DNA رنگی تحت عنوان سایبر گرین (SYBR Green) که به DNA متصل می گردد، اضافه می گردد و یا از پروب استفاده می شود.
نکته مهم: اگر ژن مورد بررسی کدینگ باشد بهتر است به عنوان فیلتر از پرایمرهای Oligo dt استفاده نماییم و اگر در مطالعه ژن های non Coding داشته باشیم یا پروکاریوت باشد(به دلیل نداشتن دم PolyA) استفاده از این پرایمرها کاربردی نیست.
کاربردها
به طور عمده بررسی بیان ژن با real time pcr یا همان PCR کمی (Quantitative PCR) انجام می شود. البته این تکنیک کاربردهای فراوانی دیگری هم چون تعیین میزان ویروس های یک نمونه، بررسی میزان تأثیر دارو درمانی، سنجش آسیب های DNA، تشخیص عوامل بیماری زا، تعیین ژنوتیپ افراد، سنجش تفکیک شدن آللی و … دارد.
نکات مهم در طراحی پرایمر
همراه RNA همیشه مقادیری gDNA داریم که ممکن است به RNA بچسپد و باعث نتایج اشتباه شود. بنابراین پرایمر ما باید بتواند RNA را از gDNA تشخیص دهد. برای تفکیک RNA از gDNA از آنزیم DNase استفاده می شود که این روش خشن محسوب می شود و ممکن است در صورت در دسترس نبودن DNA، خرد شدن RNA اتفاق بیفتد اما روش مهم برای ما، طراحی پرایمری است که RNA را از gDNA متمایز کند(RNA discrimination).
برای طراحی یک پرایمر مناسب متمایز کننده می توانیم آن را جایی طراحی کنیم که توالی روی DNA نباشد. جایی که توالی روی DNA وجود ندارد، محل اتصال دو اگزون است(Exon-Exon Junction). اما اگر منطقه اتصال بین دو اگزون به ما پرایمر خوبی نداد، از روش پُل اینترونی(Intron Spanning) استفاده می کنیم. در این روش یکی از پرایمرها را روی یک اگزون می گذاریم و دیگری را روی اگزون دیگر، نه محل اتصال دو اگزون(به عنوان مثال اگزون 1 و 4)طراحی می کنیم یا به اصطلاح پُل می زنیم.
موفقیت یا شکست در یک آزمایش PCR به پرایمر آن بستگی دارد. هدف از طراحی پرایمر پیدا کردن قطعه ای است که مکمل قطعه موردنظرمان باشد بنابراین قدم اول پبدا کردن توالی تارگت و سپس طراحی پرایمر است. پس از طراحی پرایمر با نرم افزارهای مربوط به آن، مرحله بسیار مهم آنالیز پرایمر است. آنالیز پرایمر طراحی شده ی مناسب برای تکنیک Real Time PCR نیاز به رعایت نکاتی دارد که به آنها خواهیم پرداخت.
- طول پرایمر
به طور معمول، برای یک پرایمر بهینه طول ۲۲-۱۸ نوکلئوتید را در نظر می گیرند. زیرا اگر طول پرایمر کمتر از این تعداد باشد، احتمال اتصال غیر اختصاصی بالا می رود و اگر بیشتر باشد، اتصال پرایمر به الگو در دمای Anealing سخت تر می شود.
- دمای ذوب پرایمرها(Tm)
نکته مهم بعدی در طراحی پرایمر، دمای ذوب پرایمرها یا به اصطلاح دمای Tm(دمایی که نیمی از DNA دو رشته ای از هم جدا می شود و DNA تک رشته ای ایجاد می کند) است. معمولا، پرایمرهایی با Tm بین 52 تا 58 درجه ی سانتی گراد، نتایج بهتری می دهند. در صورت بیشتر بودن دمای ذوب نسبت به نرمال رنج اتصال دو توالی سخت می شود و می تواند از تولید محصول در PCR جلوگیری کند و در صورت پایینتر بودن دمای ذوب نسبت به نرمال رنج، برخی از نوکلئوتیدهای پرایمرها به الگو متصل می شود و اتصال درستی رخ نمی دهد(جفت شدگی تحمیلی) که باعث افزایش mismatch و کاهش عملکرد اختصاصی پرایمر می شود.
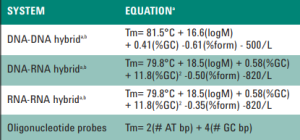
در ساده ترین و قدیمی ترین روش، دمای ذوب با استفاده از فرمول مارمور(1962) بدست می آید.
Marmur formula: Tm= 4 x GC + 2 x AT
در سال 1976 برای محاسبه دمای ذوب پرایمرها فرمول والاس مورد استفاده قرار گرفت.
Wallace formula: Tm= 64.9 + 41*(yG+zC-16.4)/(wA+xT+yG+zC)
فرمول های مارمور و والاس برای پرایمرهای دارای طول بیشتر از 13 نوکلئوتید کارایی ندارند. از سال 1986 تا کنون برای محاسبه دقیق تر Tm در پرایمرها، از فرمول پارامترهای ترمودینامیکی Breslauer(تنظیم پیش فرض Primer3) برای طراحی پرایمر استفاده می شود. با این حال، نویسندگان نرمافزار آنلاین Primer3 و سازندگان NCBI’s Primer Blast، اکنون استفاده از مجموعه پارامترهای نزدیکترین همسایه(nearest-neighbour parameter set) منتشر شده توسط SantaLucia در سال 1998 را توصیه میکنند.
- دمای اتصال پرایمر(Ta)
دمای ذوب پرایمرها(Tm) تخمینی از میزان پایداری هیبرید پرایمر با DNA الگو است و در تعیین دمای اتصال(Ta) بسیار اهمیت دارد. اگر دمای اتصال خیلی بالا باشد، پرایمرها و الگو از هم جدا باقی می مانند و اتصال صورت نمی گیرد و اگر دمای اتصال خیلی پایین باشد تنها برخی از نوکلئوتیدهای پرایمرها به الگو متصل می شود و اتصال درستی رخ نمی دهد و محصولات غیر اختصاصی تولید می شود.
از فرمول ریچلیک (Rychlik) برای به دست آوردن Ta استفاده می شود
Ta= 0.3×Tm(primer)+ 0.7×Tm(product) -14.9
بر اساس نظریه Innis و Gelfand دمای اتصال(Ta)، 3 تا 5 درجه پایینتر از دمای ذوب(Tm) می باشد.
- درصد GC
نسبت GC به کل بازهای (A=T) است که معمولا پیشنهاد میشود نرمال رنج یا درصد GC پرایمرها در بازه 45 تا 55 درصد حفظ شود.
- گیره GC یا (GC Clamp)
وجود بازهای(نوکلئوتیدهای) GC در 5 تا نوکلئوتید ابتدای پرایمر ضروری است. وجود نوکلئوتیدهای GC در بین ۵ نوکلئوتید انتهای ‘۳ پرایمر، با ایجاد پیوندهای شیمیایی پایدارتر(پیوند سه گانه)، به بهبود کارآمدی پرایمر کمک میکند. به این ترتیب، انتهای 3′ پرایمر، مانند یک گیره محکم عمل می کند و آن را به رشته الگو متصل نگه می دارد تا آنزیم پلیمراز، کار ساخت زنجیره جدید را آغاز کند. در ۵ باز انتهایی سمت ‘۳ نباید بیش از ۳ نوکلئوتید G یا C قرار داشته باشد. عدم گارانتی سر ‘۳ اتصال آنزیم Taq پلیمراز را دچار اختلال می کند.
- ساختار ثانویه پرایمر
حضور ساختارهای ثانویه ای که تحت تاثیر برهم کنش های بین مولکولی یا درون مولکولی ایجاد می شوند، منجر به تولید محصول بسیار ناچیز PCR می شود و یا اصلا محصولی تولید نمی شود(دایمر به صورت هترو و همو). وجود این ساختارهای ثانویه برخلاف اتصال پرایمرها به الگو و تکثیر عمل می کنند. این ساختارها به میزان قابل توجهی دسترسی به پرایمرها را کاهش می دهند.
نکته: پرایمرها باید برای ناحیه ای طراحی شوند که آن ناحیه ساختار ثانویه پایداری تشکیل ندهد. در غیر این صورت این ساختارهای ثانویه ی پایدار اجازه اتصال پرایمرها به الگو را نمی دهند.
- ساختارهای سنجاق سری (Hairpins)
این ساختارها جزء برهم کنش های درون مولکولی پرایمرها هستند و باید از آن ها دوری شود. برای پیش بینی ساختارهای سنجاق سری از انرژی آزاد گیپس استفاده می شود. انرژی آزاد گیبس (ΔG) انتهای ‘۳ پرایمر هرچه منفی تر باشد، ساختار پایدارتر است.
- اجتناب از همولوژی متقابل
برای بالا بردن اختصاصیت پرایمرها باید از نواحی همولوگ دوری کرد. به عبارت ساده تر، پرایمرها باید به گونه ای طراحی شوند که نواحی دیگر ژنومی را نشناسند و آن ها را تکثیر نکنند. با BLAST کردن پرایمرها این ویژگی بررسی می شود.
- طول قطعه تکثیر شونده
طول قطعه تکثیر شونده در qPCR نزدیک به ۱۰۰جفت باز و در PCR استاندارد نزدیک به ۵۰۰ جفت باز است. اگر جایگاه قرارگیری پرایمرها بر روی الگو را بدانید، طول محصول از فرمول زیر به دست می آید: طول محصول= (جایگاه پرایمر جلویی- جایگاه پرایمر عقبی)+۱
- دمای ذوب جفت پرایمرها
یک جفت پرایمر(عقبی و جلویی) باید Tm نزدیک به هم داشته باشند تا بهترین کارایی را داشته باشند. اختلاف ۵ درجه سانتی گراد یا بیشتر مانع تکثیر می شود.

چرخه انجام واکنش و برنامه دمایی
تفاوت pcr و real time چیست؟ همانند PCR کیفی در تکنیک Real Time PCR نیز به طور تقریبی همان مراحل واکنش انجام می گیرد، با این تفاوت که شما فعل و انفعالات صورت گرفته در مراحل دوم و سوم واکنش را می توانید مشاهده کنید.
- یک سیکل اختیاری 2 دقیقه ای در دمای 50 درجه سانتیگراد
این سیکل زمانی استفاده می شود که آنزیم UNG (Uracil N-glycosylase) برای حذف آلودگی های قبلی استفاده شده و این زمان باعث غیر فعال کردن آنزیم UNG در واکنش جدید می شود.
- یک سیکل 2 تا 15 دقیقه ای در دمای 90 درجه سانتیگراد
این سیکل برای فعال کردن آنزیم پلی مراز و واسرشته کردن قطعه بر اساس استفاده از نوع آنزیم پلی مراز و اندازه ی قطعه ی مورد نظر متغیر است.
- سیکل مرحله اصلی PCR (به صورت 30 تا 50 تکرار)
حالت PCR دو پله ای: 15 ثانیه در 95 درجه، اتصال و طویل سازی 60 ثانیه در 50-60 درجه یا 5 تا 8 درجه پایین تر از کمترین Tm پرایمر.
حالت PCR سه پله ای: 15 ثانیه در 95درجه، 30 ثانیه در دمای اتصال، طویل سازی 20 ثانیه در 72 درجه.
برای محاسبه مدت زمان طویل سازی از نظر تئوری برای هر 20 جفت باز با آنزیم Taq پلیمراز 1 ثانیه زمان لازم است. مرحله طویل سازی حداقل 10 ثانیه باید طول بکشد(برای محصول بالاتر از 500 جفت باز حداقل 30 ثانیه زمان لازم می باشد).
انواع روش ها
تکنیک Real Time PCR بر مبنای مولکولی که برای تشخیص استفاده می شود به دو روش تقسیم بندی می شود:
1-تشخیص غیر اختصاصی با استفاده از رنگ های باند شده به DNA(روش سایبر گرین)
در اینجا از رنگ های باند شده به DNA به عنوان گزارشگر فلوروسنت برای مشاهده واکنش PCR استفاده می شود. این فلوروسنت در سیکل متوالی بر اثر مضاعف شدن افزایش می یابد. با ثبت مقدار فلوروسنت ساطع شده در سیکل، می توان واکنش را در طول مرحله نمایی مشاهده نمود. اگر نموداری میان لگاریتم مقدار شروع واکنش و افزایش فلوروسنت گزارشگر ترسیم شود یک رابطه خطی مشاهده خواهد شد.
اغلب از سایبر گرین (SYBR Green) به همراه DNA دو رشته ای به عنوان رنگ مخصوص گزارشگر استفاده می شود. این رنگ به شکاف کوچک از مارپیچ دو رشته ای DNA باند می شود. در داخل محلول، رنگ هایی که باند نشده اند، فلوروسنت خیلی کمی را نشان می دهند و فلوروسنت زمانی به وضوح افزایش می یابد که رنگ به DNA دو رشته ای پیوسته شود.
SYBR Green تحت شرایط PCR پایدار و با ثبات باقی می ماند. سطح مطلوبی از درجه حرارت موجب تنظیم القاء و نشر طول موج ها می شود. همان طور که پیشتر ذکر شد از اتیدیوم بروماید نیز برای تشخیص می توان استفاده کرد ولی به علت سرطان زا بودن آن کمتر استفاده می شود.
2-تشخیص اختصاصی با استفاده از شناساگرهای هدف(روش Taqman)
تکنیک دیگر استفاده از پروب گزارشگر(reporter probe) یا Taqman است که با اتصال به محصولات PCR سیگنال فلوئوروسنت ایجاد میکند و استفاده از آن معمول تر است. این تکنیک به Taq Manˊ 5 nuclease assay معروف است. پروب به کار رفته در این فرایند از آن جهت reporter نامیده شده که نشانگر وجود محصول موردنظر است. محل اتصال پروب، به رشته الگو و نزدیک به پرایمرهای اصلی PCR و پایینتر از آن است.
این متد کمتر در معرض خطاهایی قرار دارد که از اتصالات نادرست از جمله دایمرهای پرایمر حاصل میشود. در این حالت هر مولکول پروب متشکل از یک الیگونوکلئوتید و دو لیبل است. رنگ فلوئوروسنت به انتهای َ۵ رشته الیگونوکلئوتیدی و ترکیب quencher به انتهای دیگر رشته متصل میشود. این ترکیب وظیفه مهار سیگنال فلوئوروسنت را بر عهده دارد و این کار را با جذب انرژی از رنگ فلوئوروسنت انجام میدهد. این پدیدهFRET (Fluorescence Resonance Energy Transfer) نام دارد.
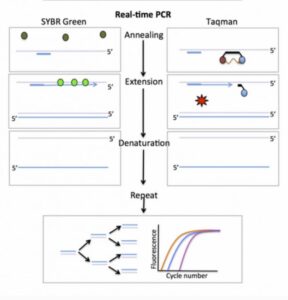
در حالت عادی سیگنال فلوئوروسنتی ایجاد نمیشود؛ چون الیگونوکلئوتید به گونه ای طراحی شده است که دو انتهای آن با همدیگر مکمل شده و رنگ فلوئوروسنت و ترکیب خاموش کننده(quencher) در کنار همدیگر قرار میگیرند. هیبریداسیون بین الیگونوکلئوتید و محصول PCR، اتصال بین دوسر الیگونوکلئوتید را بر هم میزند و این کار باعث ایجاد سیگنال فلوئوروسنت میگردد.
در حین انجام شدن PCR، پلیمراز Taq، با رسیدن به پروب با خاصیت اگزونوکلئازی ۳َ به ۵َ خود آن را تجزیه میکند و با این کار رنگ و ترکیب quencher در محیط آزاد میگردند. هر چه محصولات تولید شده بیشتر باشند، پروبهای بیشتری به توالی مکمل خود در این محصولات متصل میشوند. هر چه تعداد پروب بیشتری متصل شود، سیگنال فلوئوروسنت بیشتری حین آزاد شدن رنگ از quencher ایجاد میگردد. درنتیجه ارتباطی میان سیگنالهای فلوئوروسنت ایجاد شده در Real-time PCR و مقدار توالی الگو وجود دارد.
مراحل آزمایش
1- مرحله اول واسرشته شدن الگوی اولیه و نیز در صورت استفاده، فعال سازی آنزیم DNA پلی مراز (Hot start holding stage).
2- مرحله دوم تکنیک Real Time PCR واسرشته شدن، اتصال پرایمر الگو به هدف و تکثیر(می توان این دو مرحله را در 2 پله انجام داد، به این معنا که دمای اتصال و تکثیر را در یک بازه زمانی و یک مرحله انجام دهیم. توصیه می شود که اگر دمای اتصال پرایمر شما زیر 60 درجه باشد، این کار را انجام دهید).
مزیت PCR دو پله ای نسبت به سه پله ای، زمانی است که قطعه محصول کوتاه با دمای اتصال پائینی دارید یا دارای پرایمر چند حالته هستید. برای افزایش بازده، سرعت رمپ(سرعت کاهش یا افزایش دما در واحد زمان) را می توان افزایش داد(برای زودتر رسیدن به مرحله ی طویل سازی در دمای 72 درجه)، اما افزایش سرعت رمپ دستگاه می تواند منجر به افزایش تولید محصولات غیر اختصاصی شود. برای کاهش این اثر، زمانی که دمای اتصال پایین تر از 60 درجه باشد می توان با حذف پله سوم و استفاده از توانایی عملکرد DNA پلیمراز در دمای 55 تا 75 درجه سانتیگراد مرحله اتصال و طویل سازی را در یک پله انجام دهید.
دلیل انجام PCR سه پله ای در دمای اتصال بالای 60 درجه هم به علت این که با توجه به بهترین عملکرد آنزیم DNA پلی مراز در دمای 72 درجه سانتیگراد است. اگر دارای پرایمری با دمای اتصال 62 درجه باشید به علت فاصله دمایی کم تا 72 درجه نیاز به افزایش سرعت رمپ نیست و می توان از بهترین عملکرد آنزیم پلیمراز در دمای 72 درجه سانتی گراد استفاده کرد .
یک مرحله اختیاری: زمانی که تکثیر دایمر پرایمر نیز دارید و می خواهید داده های آن را در محاسبات نیاورید می توانید در PCR سه مرحله ای یک مرحله جمع آوری اطلاعات 15 ثانیه ای بعداز مرحله طویل سازی در دمایی که بالاتر از Tm دایمر پرایمر باشد و 3 درجه پایین تر از Tm محصول اصلی PCR است اضافه کنید. این روش باعث افزایش دامنه و اطمینان بیشتر به داده هایی که حاوی دایمر پرایمر در تکثیر بوده اند می شود. در واقع در دمای بین Tm دایمر پرایمر و Tm محصرل اصلي قطعات دايمر به صورت تک رشته ای در آمده فلورسنس آزاد نمی شود و دیگر میزان آن ها جزو داده های اصلی محاسبه نمی شود .
3- مرحله ی سوم تکنیک Real Time PCR، منحنی آنالیز ذوب که در مورد رنگ های آزاد انجام می گیرد و این مرحله جزو قابلیت های دستگاه ریل تایم پی سی آر (Real Time PCR) نسبت به دستگاه ترموسایکلر PCR معمولی است.
این پروسه شامل یک مرحله دناتوراسیون 95 درجه به مدت 15 ثانیه که دلیل انجام این کار اطمینان از انجام پروسه بر روی قطعه های اختصاصی مورد نظر است سپس گرما دهی دقیق به قطعه ی DNA تکثیر یافته از دمای 95 درجه به 60 درجه انجام می شود همان طور که گفته شد رنگ های آزاد مثل سایبرگرین که رنگ های درج شونده یا اینترکاله(Intercalating) گفته می شوند زمانی که بین دو رشته DNA قرار می گیرند و بر اثر دما دو رشته ی DNA از هم باز می شوند.
رنگ های بین رشته ای آزاد می شود که همین باعث کاهش شدت فلورسانس آن ها می شود که به صورت پیک فلورسانس بر روی نمودار آنالیز ذوب مشاهده می شوند .در آغاز آنالیز افزایش سطح فلورسانس در نمونه به علت وجود میلیون ها کپی از قطعه های تولیدی می باشد، اما هر چه نمونه گرم تر می شود و دو رشته از هم باز می شود، میزان دو رشته ای DNA کاهش یافته و در نتیجه میزان فلورسانس کاهش پیدا می کند.
دستگاه دارای یک دوربین است که این پروسه را به وسیله ی اندازه گیری فلورسانس تماشا می کند، دستگاه سپس به سادگی اطلاعات را به صورت گراف به عنوان منحنی ذوب(Melting curve) یا میزان فلورسانس در مقابل درجه حرارت را نشان می دهد. دمای ذوب دو رشته DNA در حال باز شدن از هم کاملا قابل پیش بینی است که بستگی به توالی بازهای DNA (طول و میزان GC) دارد.
بعد از اتمام واکنش PCR دستگاه Real Time به شما یک منحنی تکثیر می دهد که در بیشتر دستگاه ها به طور پیش فرض منحنی تکثیر به صورت محورهای سیکل بر فلورسانس Rn یا ΔRn نمایش داده می شود ،Rn فلورسانس خام ناشی از گزارشگر تقسیم بر فلورسانس رنگ مرجع می باشد که سیگنال گزارشگر نرمال شدهRn(Normalized Reporter signal) نامیده می شود و اگر Rn را از فلورسانس پس زمینه کم کنید، ΔRn حاصل می شود.
منحنی تکثیر
دستگاه Real-time PCR با خاصیت فلورسنتی سایبر گرین(SYBR Green) و یا پروب برای هر نمونه یک گراف یا منحنی تکثیر ترسیم می کند که هر نقطه از این گراف نشان دهنده میزان اتصال سایبر گرین و یا پروب به رشته DNA می باشد و در نهایت گراف که در نمایشگر متصل شده به Real-time PCR نشان داده می شود، به حد آستانه خود می رسد و این میزان نشان دهنده میزان DNA بیان شده در هر نمونه می باشد. تکثیر توالی DNA هدف به صورت نمایی می باشد به این صورت که یک الگو به دو، دو الگو به چهار و چهار الگو به هشت الگو و …. تبدیل میشود و این روند ادامه پیدا میکند.
در ابتدای تکثیر این موضوع درست به نظر می رسد. اما، تکثیر نمایی تا ابد ادامه نمی یابد و به طور معمول تا چرخه 35 سرعت واکنش کاهش پیدا می کند زیرا مقدار پرایمرها و dNTP ها کم می شود و بازده DNA پلیمراز نیز کم شده است، بنابراین دناتوراسیون DNA ها به طور کامل انجام نمی شود و محصولات واکنش توسط خاصیت نوکلئازی پلیمراز از بین میروند، در نتیجه واکنش وارد یک فاز خطی میشود. در این حالت مانند فاز لگاریتمی توالیهای الگو به طور کامل دو برابر نمیشوند و بازده دیگر 100% نیست. تا اینکه درنهایت تا چرخه ۴۰ام، واکنش وارد مرحله کف ثابت یا پلاتو میشود که توقف تکثیر اتفاق میافتد.
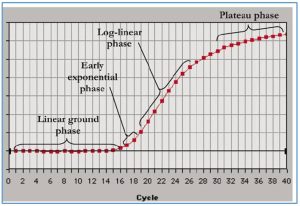
اگر منحنی تکثیر محصول در Real-time PCR را بر حسب سیکل ها ترسیم کنیم، یک منحنی که شامل 4 فاز می باشد ایجاد می شود که عبارت است از:
فاز پایه یا شروع(Baseline region / Linear ground phase): در این فاز میزان تکثیر محصولات به میزانی نیست که باعث افزایش نور فلورسنت شود. نور فلورسنت مشاهده شده در این فاز از cDNA، پرایمرها و رنگ های فلورسنت آزاد در محیط واکنش منتشر می شود به همین دلیل در این فاز میزان نور ساطع شده قابل ردیابی نیست و به صورت خطی می باشد .
فاز نمایی(Exponential phase): در این فاز میزان محصول دو رشته ای در هر چرخه دو برابر می شود بنابراین میزان محصولات به صورت نمایی افزایش پیدا می کند.
فاز ورورد به فاز خطی(Log linear phase): ترکیبات واکنش و کارایی آن ها رو به اتمام است.
فاز خطی یا مسطح(plateau phase): در این فاز، ترکیبات واکنش از بین می روند و دیگر محصولی تکثیر نمی شود به همین خاطر در میزان فلورسنت هیچ افزایشی دیده نمی شود و به اصطلاح ریل تایم در این مرحله اشباع شده است.
بررسی سیکل مرجع و خط آستانه
معمولا به صورت توافقی سیکلی که محصولات در آن وارد فاز Exponential می شوند و سیگنال واقعی از noise پس زمینه قابل تشخیص است را سیکل مرجع(cycle threshold) یا CT نامیده میشود.
مرحله ی نمایی یا Exponential phase مرحله مهم در RealTime PCR که در طول این مرحله تعیین مقدار DNA هدف امکانپذیر است مهم ترین اصطلاح در این نوع PCR اصطلاح حد آستانه است که سیکلی از PCR می باشد که میزان فلوئورسانت محصول از حد آستانه ی انتخابی ما در بالای فلورسنس پس زمینه بالاتر می رود که در این نقطه فاز نمایی آغاز می شود .
روشن است که در PCR اگر تمام شرایط برابر باشد و مهار کننده ای وجود نداشته باشد هر چه میزان نوکلئیک اسید اولیه به عنوان الگو بیشتر باشد میزان محصول و سرعت تشخیص آن ها نیز افزایش می یابد. پس در نتیجه هر چقدر میزان الگو اولیه بیشتر باشد زودتر به حد آستانه خواهیم رسید و CT کمتر خواهد شد. در یک واکنش RealTime PCR با بازده ۱۰۰٪ میزان محصولات در هر چرخه دو برابر میشود.
به این ترتیب میتوان این رابطه را نوشت:
Δ CT= 2-ΔCT fold differences
تفسیر فرمول به این صورت است که هر یک عدد کاهش در CT یعنی تعداد مولکول های آغازگر دو برابر و هر 2 عدد کاهش یعنی تعداد مولکول های هدف 4 برابر است و به همین ترتیب ادامه خواهد داشت. مقدار CT توسط نرم افزار مختص دستگاه Real Time به صورت اتوماتیک به ما داده می شود. البته به صورت دستی و با تعیین خط آستانه می توانیم آن را بررسی کنیم.
به خاطر خطاهای متعددی که حین پروسه انجام Real Time PCR رخ می دهد نظیر خطا در پیپت کردن(سمپلینگ) مواد واکنش و یا بازده و کارایی متفاوت PCR و یا حتی خطا دستگاه Real-time PCR و …که می تواند باعث ایجاد تغییر در نتایج شود. علاوه بر آن ناپایداری(Instability) مواد اولیه هم میتوانند باعث ایجاد نتایجی متغیر شوند. در نتیجه Real-time PCR، به استفاده گسترده از کنترلهایی نیازمند است که برای پی بردن به درستی نتایج حاصلشده به کار می روند.
کنترل های واکنش
یک واکنش RealTime PCR دارای چندین کنترل است
- کنترل بدون الگو یا no-template control (NTC)
در این حالت تمامی پرایمرها، پروب ها و مواد مورد نیاز جهت انجام Real-time PCR حضور دارند، ولی فقط DNA اولیه الگو نداریم. جهت بررسی تشکیل سیگنال و یا عدم تشکیل آن در غیاب نوکلئیکاسید هدف از NTC استفاده می کنیم. این کنترل قادر است که آلودگی و هرنوع فعل و انفعال پرایمرها و پروبها را که منجر به خدشه دارشدن نتایج شود، شناسایی کند.
- کنترل خارجی (exogenous)
یک قطعه DNA یا RNA سنتزشده است که با غلظت معینی در مخلوط واکنش قرار دارد. این کنترل میتواند به عنوان کنترل مثبت داخلی (Internal Positive Control= IPC) عمل کرده و مهار PCR و یک واکنش منفی حقیقی را از هم مجزا ند؛ به این ترتیب که قطعه موردنظر را به همراه پرایمر مخصوصش به مخلوط واکنش اضافه میکنیم.
اگر شرایط استاندارد باشد و واکنش درست انجام شود، باید هم این قطعه و هم توالی هدف Real-time PCR هر دو تکثیر شوند. درصورتیکه خود Real-time PCR نتیجه ندهد اما قطعه افزودهشده تکثیر شود، واکنش منفی حقیقی رخ داده است و از دلایل آن میتوان به عدم حضور توالی هدف Real-time PCR اشاره کرد. اگر هر دو قطعه تکثیر نشوند، PCR مهار شده است.
- کنترل داخلی (endogenous control)
این کنترل نوکلئیکاسیدی است که در مخلوط دارای DNA هدف وجود دارد و درصورت استفاده از پرایمرها و پروبهای مناسب، میتواند در آزمایش Real-time PCR تشخیص داده شود. این قطعه که به آن active reference هم گفته می شود جهت نرمالیزه کردن تفاوتهایی که در مقدار کل DNA اولیه اضافهشده به آزمایشهای PCR وجود دارد، به کار می رود.
- ژنهای Housekeeping
این ژن ها همیشه به یک میزان توسط سلول ها بیان می شوند و جهت تداوم بقای سلول و انجام فرایندهای حیاتی ضروری اند و در بررسی بیان ژن توسط RealTime PCR کاربرد فراوانی دارند. نظیر Albumin, Cyclophilin, β-actin ,Tubulin, GAPDH و 18S RNA

بررسی عوامل موثر بر اشکالات احتمالی منحنی تکثیر
در RealTime-PCR زمانی که منحنی تکثیری دیده نمی شود یا منحنی تکثیر عجیب و غریبی دیده شود به اولین چیزی که می توان شک کرد این است که Data Collection خاموش باشد یا فقط اطلاعات مرحله ی دوم و سوم ذخیره شده است.
دومین کاری که می توان انجام داد بررسی انتخاب صحیح کانال های رنگ است مثلا چنانچه رنگ TAMRA را برای پروب FAM انتخاب کرد بیشتر منحنی در زیر خط آستانه قرار می گیرد و نتیجه ی ضعیفی را نشان می دهد در حالی که تصحیح انتخاب درست کانال FAM باعث نمایان شدن منحنی تکثیر واقعی می شود و می توان از طریق بررسی و انطباق منحنی حاصل از تابش خام (در وضعیت Raw spectra) و منحنی Best fit اشتباه در انتخاب کردن کانال های رنگ را تشخیص داد که در صورت عدم انطباق این دو منحنی به انتخاب رنگ اشتباه و یا آلودگی یا رنگ دیگر در مخلوط واکنش پی برد.
زمانی که برای دستگاه سهوا چاهک بدون نمونه و خالی را به عنوان نمونه مورد آنالیز برای تعریف کنید، منحنی تکثیری که دستگاه به ما نشان می دهد، به صورت نامنظم با آستانه ی غیر منتظره می باشد (ظرف خالی نیز دارای مقدار بسیار کمی فلورسنس می باشد ). زمانی که از بلانک آب و یا از ROX به عنوان رنگ مرجع قرار است استفاده کنید، اما در مخلوط واکنش ROX وجود نداشته باشد نیز منحنی تکثیری را مشاهده می کنید.
حد پایه
بعد از داشتن یک منحنی تکثیر با 4 فاز استاندارد، نوبت به تنظیم حد پایه Baseline می رسد. حد پایه نویز در سیکل های اول است که به طور معمول بین سیکل های 3 تا 15 تعیین می شود، جایی که فلورسنس قابل تشخیص به وسیله ی دستگاه نیست.
تعیین محل سیکل های مورد استفاده برای حساب کردن حد پایه می تواند تغییر کند و در مقادیر الگوی بالا یا اگر میزان بیان خیلی بالا باشد، نیاز به کاهش حد پایه می باشد. برای تصحیح حد پایه، اطلاعات فلورسنس را در گراف منحنی تکثیر با مقیاس خطی (به جای مقیاس لگاریتمی ) مشاهده کرده و حد پایه در محدوده ی بالا را در جایی که کم ترین Ct شروع شده، قرار می دهیم.
برای مثال اگر کم ترین Ct برابر با 15 باشد حد پایه که در محدوده 6 تا 15 قرار داشته را در محدوده ی 6 تا 13 قرار می دهیم ( قانون کلی تعیین حد پایه ،2 تا 3 سیکل کمتر از کمترین Ct را به عنوان سطح بالای حد پایه تعیین کنید).
حد آستانه
بعد از انتخاب صحیح Baseline در منحنی تکثیر،نوبت به انتخاب آستانه می رسد. آستانه به مرز بالای فلورسنس پس زمینه گفته می شود که در زیر فاز سکون از منحنی تکثیر قرار دارد. مقدار آستانه باید در فاز لگاریتمی منحنی تنظیم شود تا به آسانی در فاز لگاریتمی و فاز خطی قابل تشخیص باشد. اگر چندین هدف را بررسی می کنید ،باید آستانه را برای هر کدام به طور جداگانه تنظیم کنید. دستگاه به طور خودکار آستانه را 10 برابر SD حد پایه تنظیم می کند.
اگر بخواهیم به طور دستی آستانه را تنظیم کنیم
1) آستانه باید در جایی از منحنی تکثیر باشد که فاز لگاریتمی همه نماهای منحنی را در بر بگیرد، نباید آستانه در فاز سکون و یا در اواخر فاز خطی باشد.
2) آستانه باید در جایی از منحنی تکثیر قرار بگیرد، که حداکثر دقت تکثیر وجود دارد و تکثیر به درستی در همه ی نماهای منحنی تکثیر انجام شده باشد، که نشان دهنده ی بالاترین حساسیت تست باشد.
منحنی ذوب
بررسی کنترل ها و نمونه اصلی
کنترل مثبت: به جهت اطمینان از درست انجام شدن واکنش و نیز سالم بودن مخلوط واکنش از کنترل مثبت استفاده می شود. می توانید از نمونه ی بیمار مثبت یا نمونه ای که از داشتن هدف مورد نظرتان اطمینان دارید، استفاده کنید. یکی از بهترین مواردی که می توانید به عنوان کنترل مثبت استفاده کنید، منحنی استاندارد می باشد.
کنترل منفی یا کنترل فاقد الگو (NTCs): به منظور بررسی آلودگی مخلوط واکنش در آزمایش مورد بررسی قرار می گیرد و مثبت شدن آن نشان از آلودگی مخلوط واکنش دارد و نمی توان به داده های حاصل از نمونه های اصلی اعتماد کرد. البته در مواردی که حل مشکل آلودگی ممکن نباشد، می توان با بررسی نتایج نیز جواب قابل اعتمادی به دست آورد.
به این صورت که اگر CT کنترل NTC در سیکل 35 تا 40 قرار بگیرد و Ct نمونه اصلی در سیکل 25 یا کمتر بود ( در واقع اختلاف 10 سیکل بین نمونه ی اصلی و کنترل NTC در صورتی کهCT کنترل بین 35 تا 40 باشد ) می توان به جواب نمونه ی اصلی اعتماد کرد. در صورت قرار گرفتن Ct کنترل NTC در سیکل های پایین تر از 30، به احتمال زیاد آلودگی در مخلوط واکنش وجود دارد و نمی توان به جواب آزمایش اعتماد کرد .
نمونه اصلی: بعد از تنظیم و بررسی مراحل بالا، نوبت به بررسی نمونه های اصلی مورد آنالیز می باشد. نمونه های اصلی نیز باید دارای منحنی تکثیر مناسب باشد، تکثیر باید به صورت واقعی صورت بگیرد، که در این صورت میزان ΔRn در فاز سکون منحنی تکثیر باید بیشتر از 0/5 باشد. اگر تکثیری وجود نداشت، فلورسانس پروب یا پرایمر یا بررسی کنید.
اگر فلورسانس وجود داشت، پروب سالم می باشد و احتمالا الگوی واکنش از بین رفته یا اصلا در مخلوط واکنش الگو اضافه نکرده اید. اگر فلورسانس وجود نداشت، کلا بررسی نمونه زیر سؤال است و آن را از داده هایتان حذف کنید. نباید اختلاف CT بین تکرار نمونه های حداکثر بیشتر از 0/5باشد (در هر عملیات واکنشPCRهر نمونه مورد آنالیز را باید 3 بار تکرار کنید)، که در این صورت دارای ضریب تغییرات (CV) کمی نسبت به هم خواهند بود. اگر در بین تکرار ها، نمونه ای دارای Ct کمتر یا بیشتر بود، می توان به میزان الگویی که اضافه کرده اید شک کنید، دو برابر بودن الگو میزان Ct را یک سیکل کاهش می دهد .
اگر CT نمونه های اصلی دو سیکل های 35 تا 39 قرار بگیرد، برای اطمینان به این که سیگنال نمونه بالاتر از پس زمینه قرار می گیرد و سیگنال ناشی از آلودگی یا مسائل دیگر نیست، دو کار می توانید انجام دهید؛ یا الگوی مخلوط واکنش را دو برابر کنید که در این صورت باید CT واکنش یک سیکل کم تر شود، یا تعداد سیکل های واکنش را به 50 سیکل افزایش دهید تا اختلاف 10 سیکل بین نمونه ی اصلی و نمونه ی کنترل بدون الگو مشاهده شود و اگر این اختلاف مشاهده نشد، نشان از آلودگی می باشد.
آنالیز منحنی ذوب
در روش استفاده از رنگ های آزاد، برای بررسی عملکرد پرایمر های طراحی شده و اطمینان از این که پرایمر فقط به هدف مورد نظر متصل شده و تکثیر به درستی و با اختصاصیت کافی انجام شده است، می توانید با بررسی آنالیز ذوب نتایج را تفسیر کنید، در واقع وجود منحنی تکثیر فقط می تواند نشان از یک تکثیر باشد و واقعی بودن یا نبودن اختصاصیت آن باید تأیید شود، که می توان از منحنی آنالیز ذوب استفاده کرد. همان طور که گفته شد، افت فلورسنس بر روی منحنی نمودار منحنی ذوب به صورت قله دیده می شود که در واقع برای مشاهده ی راحت تر نمودار این چنین نمایش داده می شود.
برای درک بهتر یک مثال برایتان خواهیم گفت، اگر اندازه ی قطعه 130 جفت باز و دمای Tm محصول PCR ما 80 درجه سانتی گراد باشد این انتظار را داریم که در منحنی آنالیز ذوب فقط یک پیک فلورسانس در محل دمای Tm که 80 درجه است داشته باشیم.
اگر دو پیک فلورسانس در منحنی آنالیز ذوب مشاهده شد، نشان از تکثیر دو قطعه ی تولیدی می باشد و پرایمر طراحی شده به صورت غیر اختصاصی به یک توالی دیگر متصل شده است. احتمال زیاد اگر پیک دوم در دمای 75 درجه سانتی گراد مشاهده شد، نشان از تشکیل دایمر پرایمر و غیر بهینه بودن واکنش دارد. توجه داشته باشید که در هنگام طراحی پرایمر، نرم افزار به شما دمای ذوب قطعه ی تولیدی را خواهد داد و شما می توانید بر اساس آن منحنی آنالیز ذوب خود را بررسی و آنالیز کنید.









نقد و بررسیها
هنوز بررسیای ثبت نشده است.