


Blog
Real time PCR
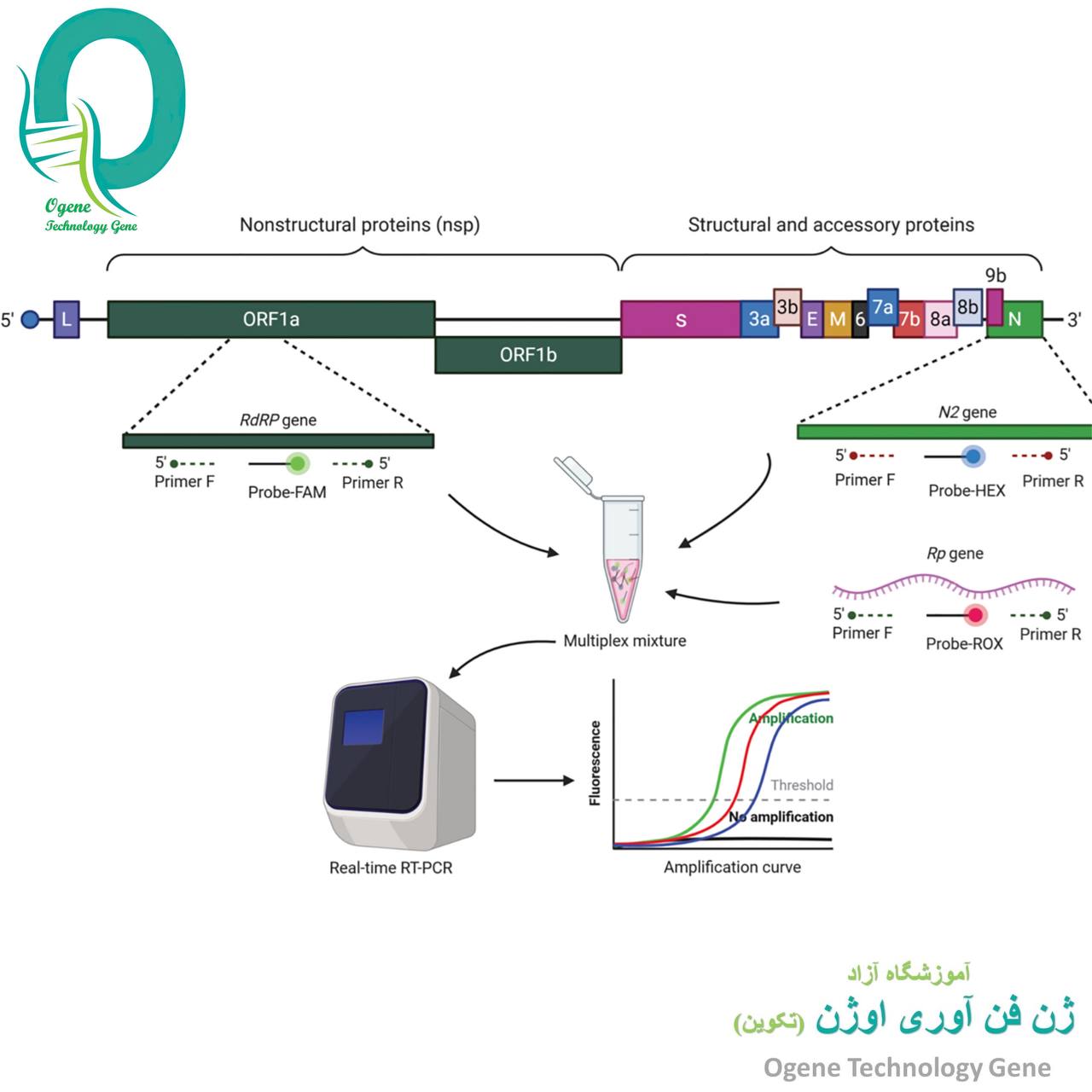
real time PCR چیست؟
تکنیک Real Time PCR(ریل تایم پی سی آر) که به آن Quantitative PCR یا PCR کمی نیز می گویند. به طور عمده بررسی بیان ژن با real time pcr انجام می شود هر چند که در تعیین میزان ویروس های یک نمونه هم تکنیکی قوی و حساس به شمار می آید، البته این تکنیک کاربردهای فراوانی دیگری هم چون بررسی میزان تأثیر دارو درمانی، سنجش آسیب های DNA، تشخیص عوامل بیماری زا، تعیین ژنوتیپ افراد، سنجش تفکیک شدن آللی و … دارد.
در تکنیک Real Time PCR از یک مولکول گزارشگر فلوروسنت برای مشاهده پیشرفت PCR استفاده می شود و قدرت سیگنال فلوئورسنت تولید شده ارتباط مستقیمی با مقدار مولکول های تکثیرشده دارد. در تکنیک Real Time PCR میزان محصولاتی که حین یک آزمایش PCR تولید می شوند با میزان محصولات تولید شده طی آزمایش های PCR با مقدار نوکلئیک اسید آغازکننده مشخص، مقایسه میشود و امکان پی بردن به مقدار نوکلئیک اسید اولیه موجود در نمونه، برای ما فراهم می شود.
در وهله اول برای انجام آزمایش real-time PCR نیاز به نمونه هایی که سنتز cDNA شده اند داریم، بنابراین قبل از فرآیند سننتز cDNA بایستی از نمونه ها، RNA استخراج کرده باشیم و RNA ای استخراج شده را مورد سنجش غلظت یا خلوص قرار دهیم(اسپکتروفتومتری) که ببینیم نمونه های RNA که استخراج شدند قابلیت این که ما بخواهیم با آنها cDNA را سنتز کنیم، دارند یا خیر و در مرحله بعد سنتز cDNA را انجام می دهیم.
برای سنتز cDNA می دانیم که اگر بخواهیم آنالیز بیان ژن انجام دهیم که این آزمایش همان استفاده از تکنیک Real Time PCR هست. برای برای انجام آزمایش آنالیز بیان ژن باید mRNA را بررسی کنیم، چون mRNA ناپایدار است، و در مدت زمان کمی از بین می رود، به همین دلیل یکی از اهدافی که cDNA انجام می دهیم این است که mRNA را از حالت ناپایدار تبدیل به فرم پایدار کنیم تا مدت زمان نگهداری mRNA بیشتر شود. اما هدف اصلی ما از انجام real time این است که، به یک DNA پلی مرازی نیاز داریم RNA را به DNA تبدیل کنیم.
از دیگر مواد مهم برای سنتز cDNA، پرایمرها هستند که برای این کار 3 نوع پرایمر وجود دارد:
- پرایمرهای رندوم هگزامر (Random Hexamer): توالی های کوچک 6 نوکلئوتیدی که به صورت تصادفی به هر جایی از RNA امکان اتصال دارند. این پرایمرها زمانی که بخواهیم بیان تمام ژن ها را تحت هر شرایطی با هر ژن کنترلی حتی 18s rRNA بررسی کنیم کاربرد دارد.
- پرایمرهای OligodT: این پرایمرها به انتهای mRNA دارای توالی polyA متصل می شوند.
- پرایمرهای اختصاصی: این پرایمرها برای نقاط شناخته شده ای از mRNA ها با توالی مشخص طراحی می شوند و در واقع باعث بهینه سازی توالی مورد نظر می گردند و در کلونینگ کاربرد دارند. مواد دیگری که در سنتز cDNA نقش دارند شامل نوکلئوتیدها( dNTPها)، بافر آنزیم RT، آنزیم ریبونوکلئاز و RNase H که وظیفه ی جداسازی دو رشته ی RNA و cDNA سنتز شده از هم را دارد، می باشد. که معمولا همه این مواد به صورت مخلوطی در کیت های سنتز cDNA در ویالی تحت عنوان Master Mix موجود می باشد و نیازی به افزودن آن ها به صورت جداگانه نیست.
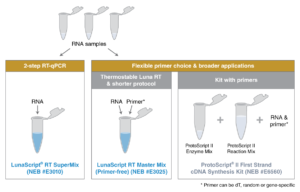
سنتز cDNA مرحله مهم در تکنیک Real Time PCR است که برای انجام این آزمایش، برخی کیت ها به صورت One step و برخی Two step می باشند.
در حالت Two step ابتدا سنتز cDNA در یک ویال انجام می شود و سپس PCR معمولی در جهت بررسی بیان ژن cDNA سنتزشده در ویالی دیگر انجام می شود. این کیت ها دقت بیشتری دارند و باعث آسانتر شدن Trouble shooting در هر مرحله می شوند.
در حالت One step همه ی مراحل سنتز cDNA تا بیان ژن در یک ویال انجام می گیرد و برای افرادی که تسلط زیادی دارند مناسب تر است. برای انجام این تکنیک دستهها ابتدا استخراج RNAموجود در نمونه مورد نظر انجام می شود. RNA استخراج شده جهت سنتز cDNA باید کیفیت و غلظت مناسبی داشته باشد چون چیزی معادل 1μg یا همان 1000ng از RNA در هر بار استفاده جهت سنتز مورد نیاز است.
سپس RNA مورد نظر را با موادی که در بالا گفته شد بر روی یخ مخلوط کردیم ویال را به دستگاه ترموسایکلر منتقل می کنیم تا دماهای لازم را با برنامه زمانی که بسته به کیت متفاوت است اعمال کنیم.
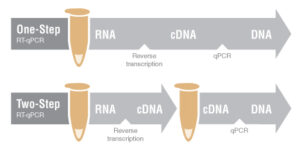
موفقیت یا شکست در یک واکنش PCR به پرایمر آن بستگی دارد. برای طراحی پرایمر مناسب تکنیک Real Time PCR لازم به رعایت نکاتی هستیم، که شامل موارد زیر می شود:
1- طول پرایمر:
به طور معمول، برای یک پرایمر بهینه طول ۲۲-۱۸ نوکلئوتید را در نظر می گیرند. زیرا اگر طول پرایمر کمتر از این تعداد باشد، احتمال اتصال غیر اختصاصی بالا می رود و اگر بیشتر باشد، اتصال پرایمر به الگو در دمای Anealing سخت تر می شود.
2- دمای ذوب پرایمرها:
نکته مهم بعدی در طراحی پرایمر، دمای ذوب پرایمرها یا به اصطلاحTm، دمایی است که نیمی از DNA دو رشته ای از هم جدا می شود و DNA تک رشته ای ایجاد می کند. معمولا، پرایمرهایی با Tm بین 52 تا 58 درجه ی سانتی گراد، نتایج بهتری را در PCR به دست می دهند. برخی از نوکلئوتیدهای پرایمرها به الگو متصل می شود و اتصال درستی رخ نمی دهد و محصولات غیر اختصاصی تولید می شود.
از فرمول ریچلیک (Rychlik) برای به دست آوردن Ta استفاده می شود:
Ta= 0.3×Tm(primer)+ 0.7×Tm(product) -14.9
3- دمای اتصال (Anealing) پرایمر:
دمای ذوب پرایمرها تخمینی از میزان پایداری هیبرید DNA-DNA است و در تعیین دمای اتصال خیلی اهمیت دارد. اگر دمای اتصال (Ta) خیلی بالا باشد، پرایمرها و الگو از هم جدا باقی می مانند و اتصال صورت نمی گیرد و اگر دمای اتصال خیلی پایین باشد تنها برخی از نوکلئوتیدهای پرایمرها به الگو متصل می شود و اتصال درستی رخ نمی دهد و محصولات غیر اختصاصی تولید می شود.
4- درصد GC
نسبت GC به کل بازهای A=T است که معمولا پیشنهاد میشود درصد GC پرایمرها در بازه 40 تا 60 درصد حفظ شود.
5- گیره GC (clamp)
وجود بازهای(نوکلئوتیدهای) GC در 5 تا نوکلئوتید ابتدای پرایمر ضروری است.
وجود نوکلئوتیدهای G C در بین ۵ نوکلئوتید انتهای 3′ پرایمر، با ایجاد پیوندهای شیمیایی پایدارتر )سه گانه) ، به بهبود کارآمدی پرایمر کمک میکند. به این ترتیب، انتهای 3′ پرایمر، مانند یک گیره محکم عمل می کند و آن را به رشته الگو متصل نگه می دارد تا آنزیم پلیمراز، کار ساخت زنجیره جدید را آغاز کند. در ۵ باز انتهایی سمت ‘۳ نباید بیش از سه G یا C قرار داشته باشد.
6- ساختار ثانویه پرایمر:
حضور ساختارهای ثانویه ای که تحت تاثیر برهم کنش های بین مولکولی یا درون مولکولی ایجاد می شوند، منجر به تولید محصول بسیار ناچیز PCR می شود و یا اصلا محصولی تولید نمی شود. وجود این ساختارهای ثانویه برخلاف اتصال پرایمرها به الگو و تکثیر عمل می کنند. این ساختارها به میزان قابل توجهی دسترسی به پرایمرها را کاهش می دهند.
نکته: پرایمرها باید برای ناحیه ای طراحی شوند که آن ناحیه ساختار ثانویه پایداری تشکیل ندهد. در غیر این صورت این ساختارهای ثانویه ی پایدار اجازه اتصال پرایمرها به الگو را نمی دهند.
7- ساختارهای سنجاق سری (hairpins) :
این ساختارها جزء برهم کنش های درون مولکولی پرایمرها هستند و باید از آن ها دوری شود. انرژی آزاد گیبس (ΔG) انتهای ‘۳ پرایمر هرچه منفی تر باشد، ساختار پایدارتر است.
8- اجتناب از همولوژی متقابل :
برای بالا بردن اختصاصیت پرایمرها باید از نواحی همولوگ دوری کرد. به عبارت ساده تر، پرایمرها باید به گونه ای طراحی شوند که نواحی دیگر ژنومی را نشناسند و آن ها را تکثیر نکنند. با BLAST کردن پرایمرها این ویژگی بررسی می شود.
9- طول قطعه تکثیر شونده:
طول قطعه تکثیر شونده در qPCR نزدیک به ۱۰۰جفت باز و در PCR استاندارد نزدیک به ۵۰۰ جفت باز است. اگر جایگاه قرارگیری پرایمرها بر روی الگو را بدانید، طول محصول از فرمول زیر به دست می آید: طول محصول= (جایگاه پرایمر جلویی- جایگاه پرایمر عقبی)+۱
10- دمای ذوب جفت پرایمرها:
یک جفت پرایمر(عقبی و جلویی) باید Tm نزدیک به هم داشته باشند تا بهترین کارایی را داشته باشند. اختلاف ۵ درجه سانتی گراد یا بیشتر مانع تکثیر می شود.
انواع روش های real time PCR
تکنیک Real Time PCR بر مبنای مولکولی که برای تشخیص استفاده می شود به دو روش تقسیم بندی می شود:
1-تشخیص غیر اختصاصی با استفاده از رنگ های باند شده به DNA
در اینجا از رنگ های باند شده به DNA به عنوان گزارشگر فلوروسنت برای مشاهده واکنش PCR استفاده می شود. این فلوروسنت در سیکل متوالی بر اثر مضاعف شدن افزایش می یابد. با ثبت مقدار فلوروسنت ساطع شده در سیکل، می توان واکنش را در طول مرحله نمایی مشاهده نمود.
اگر نموداری میان لگاریتم مقدار شروع واکنش و افزایش فلوروسنت گزارشگر ترسیم شود یک رابطه خطی مشاهده خواهد شد. اغلب از سایبر گرین (SYBR Green) به همراه DNA دو رشته ای به عنوان رنگ مخصوص گزارشگر استفاده می شود. این رنگ به شکاف کوچک از مارپیچ دو رشته ای DNA باند می شود. در داخل محلول، رنگ هایی که باند نشده اند فلوروسنت خیلی کمی را نشان می دهند و فلوروسنت زمانی به وضوح افزایش می یابد که رنگ به DNA دو رشته ای پیوسته شود.
SYBR Green تحت شرایط PCR پایدار و با ثبات باقی می ماند. سطح مطلوبی از درجه حرارت موجب تنظیم القاء و نشر طول موج ها می شود. همان طور که پیشتر ذکر شد از اتیدیوم بروماید نیز برای تشخیص می توان استفاده کرد ولی به علت سرطان زا بودن آن کمتر استفاده می شود.
2-تشخیص اختصاصی با استفاده از شناساگرهای هدف
تکنیک دیگر استفاده از پروب گزارشگر (reporter probe) یا Taqman است که با اتصال به محصولات PCR سیگنال فلوئوروسنت ایجاد میکند و استفاده از آن معمول تر است. این تکنیک به Taq Manˊ 5 nuclease assay معروف است. پروب به کار رفته در این فرایند از آن جهت reporter نامیده شده که نشانگر وجود محصول موردنظر است. محل اتصال پروب، به رشته الگو و نزدیک به پرایمرهای اصلی PCR و پایینتر از آن است.
این متد کمتر در معرض خطاهایی قرار دارد که از اتصالات نادرست از جمله دایمرهای پرایمر حاصل میشود. در این حالت هر مولکول پروب متشکل از یک الیگونوکلئوتید و دو لیبل است. رنگ فلوئوروسنت به انتهای َ۵ رشته الیگونوکلئوتیدی و ترکیب quencher به انتهای دیگر رشته متصل میشود. این ترکیب وظیفه مهار سیگنال فلوئوروسنت را بر عهده دارد و این کار را با جذب انرژی از رنگ فلوئوروسنت انجام میدهد. این پدیده FRET (Fluorescence Resonance Energy Transfer) نام دارد.
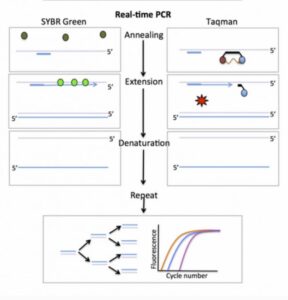
در حالت عادی سیگنال فلوئوروسنتی ایجاد نمیشود؛ چون الیگونوکلئوتید به گونه ای طراحی شده است که دو انتهای آن با همدیگر مکمل شده و رنگ فلوئوروسنت و ترکیب quencher در کنار همدیگر قرار میگیرند. هیبریداسیون بین الیگونوکلئوتید و محصول PCR، اتصال بین دوسر الیگونوکلئوتید را بر هم میزند و این کار باعث ایجاد سیگنال فلوئوروسنت میگردد.
در حین انجام شدن PCR، پلیمراز Taq، با رسیدن به پروب با خاصیت اگزونوکلئازی ۳َ به ۵َ خود آن را تجزیه میکند و با این کار رنگ و ترکیب quencher در محیط آزاد میگردند. هر چه محصولات تولید شده بیشتر باشند، پروبهای بیشتری به توالی مکمل خود در این محصولات متصل میشوند. هر چه تعداد پروب بیشتری متصل شود، سیگنال فلوئوروسنت بیشتری حین آزاد شدن رنگ از quencher ایجاد میگردد. درنتیجه ارتباطی میان سیگنالهای فلوئوروسنت ایجاد شده در Real-time PCR و مقدار توالی الگو وجود دارد.
تفاوت PCR و real time چیست؟
تکنیک Real Time PCR، چرخه انجام واکنش و برنامه دمایی:
همانند PCR کیفی در تکنیک Real Time PCR نیز به طور تقریبی همان مراحل واکنش انجام می گیرد، با این تفاوت که شما فعل و انفعالات صورت گرفته در مراحل دوم و سوم واکنش را می توانید مشاهده کنید.
1 – یک سیکل اختیاری 2 دقیقه ای در دمای 50 درجه سانتیگراد
این سیکل زمانی استفاده می شود که آنزیم UNG (Uracil N-glycosylase) برای حذف آلودگی های قبلی استفاده شده و این زمان باعث غیر فعال کردن آنزیم UNG در واکنش جدید می شود.
2- یک سیکل 2 تا 15 دقیقه ای در دمای 90 درجه سانتیگراد:
این سیکل برای فعال کردن آنزیم پلی مراز و واسرشته کردن قطعه بر اساس استفاده از نوع آنزیم پلی مراز و اندازه ی قطعه ی مورد نظر متغیر است.
3 – سیکل مرحله اصلی PCR (به صورت 30 تا 50 تکرار):
حالت PCR دو پله ای : 15 ثانیه در 95 درجه ، اتصال و طویل سازی 60 ثانیه در 50-60 درجه یا 5 تا 8 درجه پایین تر از کمترین Tm پرایمر.
حالت PCR سه پله ای : 15 ثانیه در 95درجه، 30 ثانیه در دمای اتصال، طویل سازی 20 ثانیه در 72 درجه.
برای محاسبه مدت زمان طویل سازی از نظر تئوری برای هر 20 جفت باز با آنزیم Taq پلی مراز 1 ثانیه زمان لازم است. مرحله طویل سازی
حداقل 10 ثانیه باید طول بکشد. برای محصول بالاتر از 500 جفت باز حداقل 30 ثانیه زمان لازم می باشد.

مراحل سه گانه تکنیک Real Time PCR
1- تکنیک Real Time PCR: مرحله اول واسرشته شدن الگوی اولیه و نیز در صورت استفاده فعال سازی آنزیم DNA پلی مراز Hot start holding stage .
2- مرحله دوم تکنیک Real Time PCR: در این مرحله واسرشته شدن، اتصال پرایمر الگو به هدف و تکثیر انجام می شود ( می توان این دو مرحله را در 2 پله انجام داد به این معنا که دمای اتصال و تکثیر را در یک بازه زمانی و یک مرحله انجام دهیم. توصیه می شود که اگر دمای اتصال پرایمر شما زیر 60 درجه باشد، این کار را انجام دهید).
مزیت PCR دو پله ای نسبت به سه پله ای، زمانی است که قطعه محصول کوتاه با دمای اتصال پائینی دارید یا دارای پرایمر چند حالته هستید. برای افزایش بازده، سرعت رمپ ( سرعت کاهش یا افزایش دما در واحد زمان ) را می توان افزایش داد ( برای زودتر رسیدن به مرحله ی طویل سازی در دمای 72 درجه )، اما افزایش سرعت رمپ دستگاه می تواند منجر به افزایش تولید محصولات غیر اختصاصی شود.
برای کاهش این اثر، زمانی که دمای اتصال پایین تر از 60 درجه باشد می توان با حذف پله سوم و استفاده از توانایی عملکرد DNA پلی مراز در دمای 55 تا 75 درجه سانتیگراد مرحله اتصال و طویل سازی را در یک پله انجام دهید.
دلیل انجام PCR سه پله ای در دمای اتصال بالای 60 درجه هم به علت این که با توجه به بهترین عملکرد آنزیم DNA پلی مراز در دمای 72 درجه سانتیگراد است اگر دارای پرایمری با دمای اتصال 62 درجه باشید به علت فاصله دمایی کم تا 72 درجه نیاز به افزایش سرعت رمپ نیست و می توان از بهترین عملکرد آنزیم پلی مراز در دمای 72 درجه سانتی گراد استفاده کرد .
یک مرحله اختیاری: زمانی که تکثیر دایمر پرایمر نیز دارید و می خواهید داده های آن را در محاسبه نیاورید می توانید در PCR سه مرحله ای یک مرحله جمع آوری اطلاعات 15 ثانیه ای بعداز مرحله طویل سازی در دمایی که بالاتر از Tm دایمر پرایمر باشد و 3 درجه پایین تر از Tm محصول اصلی PCR است اضافه کنید.
این روش باعث افزایش دامنه و اطمینان بیشتر به داده هایی که حاوی دایمر پرایمر در تکثیر بوده اند می شود. در واقع در دمای بین Tm دایمر پرایمر و Tm محصرل اصلي قطعات دايمر به صورت تک رشته ای در آمده فلورسنس آزاد نمی شود و دیگر میزان آن ها جزو داده های اصلی محاسبه نمی شود .
3- مرحله ی سوم تکنیک Real Time PCR، منحنی آنالیز ذوب که در مورد رنگ های آزاد انجام می گیرد و این مرحله جزو قابلیت های دستگاه ریل تایم پی سی آر (Real Time PCR) نسبت به دستگاه ترموسایکلر PCR معمولی است.
این پروسه شامل یک مرحله دناتوراسیون 95 درجه به مدت 15 ثانیه که دلیل انجام این کار اطمینان از انجام پروسه بر روی قطعه های اختصاصی مورد نظر است سپس گرما دهی دقیق به قطعه ی DNA تکثیر یافته از دمای 95 درجه به 60 درجه انجام می شود همان طور که گفته شد رنگ های آزاد مثل سایبرگرین که رنگ های درج شونده یا اینترکاله (Intercalating) گفته می شوند زمانی که بین دو رشته DNA قرار می گیرند و بر اثر دما دو رشته ی DNA از هم باز می شوند.
رنگ های بین رشته ای آزاد می شود که همین باعث کاهش شدت فلورسانس آن ها می شود که به صورت پیک فلورسانس بر روی نمودار آنالیز ذوب مشاهده می شوند .در آغاز آنالیز افزایش سطح فلورسانس در نمونه به علت وجود میلیون ها کپی از قطعه های تولیدی می باشد، اما هر چه نمونه گرم تر می شود و دو رشته از هم باز می شود، میزان دو رشته ای DNA کاهش یافته و در نتیجه میزان فلورسانس کاهش پیدا می کند.
دستگاه دارای یک دوربین است که این پروسه را به وسیله ی اندازه گیری فلورسانس تماشا می کند، دستگاه سپس به سادگی اطلاعات رت به صورت گراف به عنوان منحنی ذوب (Melting curve) میزان فلورسانس در مقابل درجه حرارت را نشان می دهد. دمای ذوب دو رشته DNA در حال باز شدن از هم کاملا قابل پیش بینی است که بستگی به توالی بازهای DNA (طول و میزان GC) دارد.
بعد از اتمام واکنش PCR دستگاه Real Time به شما یک منحنی تکثیر می دهد که در بیشتر دستگاه ها به طور پیش فرض منحنی تکثیر به صورت محورهای سیکل بر فلورسانس Rn یا ΔRn نمایش داده می شود ،Rn فلورسانس خام ناشی از گزارشگر تقسیم بر فلورسانس رنگ مرجع می باشد که سیگنال گزارشگر نرمال شده Rn (Normalized Reporter signal) نامیده می شود و اگر Rn را از فلورسانس پس زمینه کم کنید، ΔRn حاصل می شود.
- روش بررسی بیان ژن با روش ریل تایم پی سی آر (real time pcr)
تکنیک Real Time PCR بسیار شبیه به روش PCRمی باشد و در ریل تایم نیز همانند PCR با استفاده از پرایمرهای اختصاصی، یک توالی تکثیر می گردد. اما تفاوت ریل تایم با PCR معمولی در سنجش کمی توالی تکثیر شده می باشد. در روش ریل تایم با به کار گرفتن یک نشانگر فلورسنت در واکنش، میزان تکثیر محصول ردیابی می گردد. این نشانگرهای فلورسنت به گونه ای طراحی میشوند که در صورت تکثیر DNA ، با اتصال آن ها به DNA نور تولید کنند. بنابراین نور بیشتر برابر است با تکثیر محصول و افزایش شدت نور ثبت شده در دستگاه با میزان محصول بدست آمده نسبت مستقیم دارد.
- اصول real time pcr : سنجش میزان بیان ژن ها با تکنیک ریل تایم به چه صورت می باشد؟
روش ریل تایم به دو دسته تقسیم بندی می شود:
روش اول: استفاده از نشانگرهای فلورسنت غیر اختصاصی با استفاده از رنگ های باند شده بهDNA مانند (SYBR® Green) و یا اوا گرین (Eva green)
در این روش از رنگ های متصل شوند به DNA دو رشته ای، به عنوان گزارشگر فلوروسنت برای مشاهده واکنش PCR استفاده می شود. این دو رنگ به شکاف کوچک مارپیچ دو رشته ای DNA باند می شود و سبب ایجاد نور فلورسنت می گردند، اما رنگ هایی که به رشته DNA باند نشده اند فلوروسنت خیلی کمی را نشان می دهند (شکل 1).
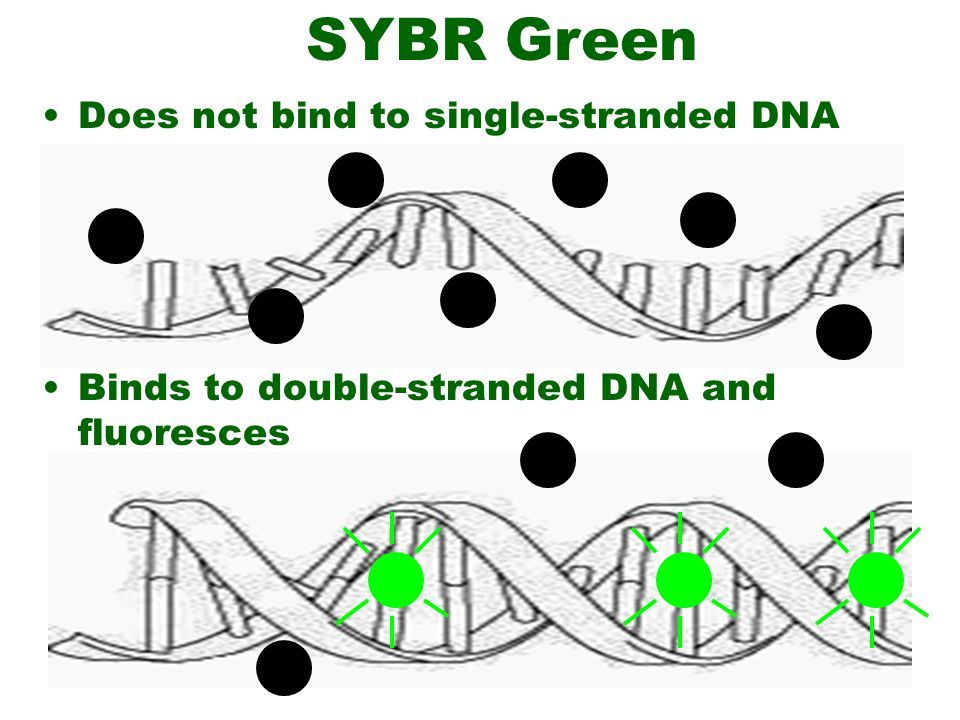
شکل 1: این شکل بیانگر قابلیت اتصال رنگ های فلورسنت به DNA می باشد.
بنابراین در روش ریل تایم، با تکثیر DNA قطعات دو رشته ای افزایش می یابند و با اتصال رنگ های فلورسنت به آن ها میزان نور انتشار شده نیز بیشتر می گردد. و این انتشار نور فلوروسنت در سیکل های متوالی بر اثر مضاعف شدن محصول PCR افزایش می یابد (شکل 2) بنابراین با ثبت مقدار فلوروسنت ساطع شده در هر سیکل، می توان پیشرفت تکثیر محصولات واکنش PCR را در طول مرحله نمایی PCR مشاهده نمود.
نکته: رنگ های سایبر گرین (SYBR® Green) و یا اوا گرین (Eva green) قابلیت اتصال به DNA تک رشته را ندارند.
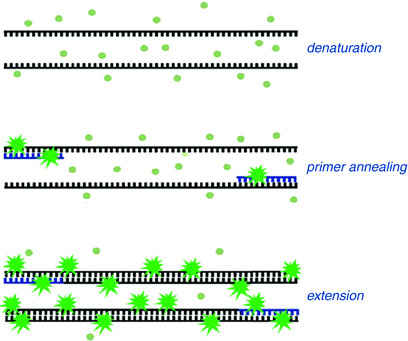
شکل 2: عملکرد رنگ های فلورسنت را در اتصال به DNA دورشته ای در روش ریل تایم.
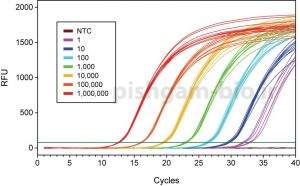
شکل 3: نمودار تکثیر محصولات در ریل تایم بر حسب سیکل.
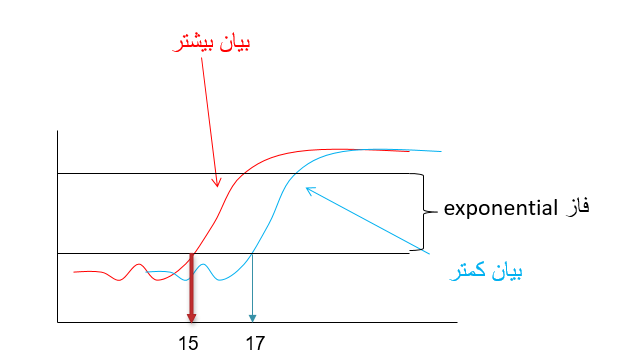
شکل 4: این شکل منحنی تکثیر ژن YHZ2 را در دو نمونه مختلف (به عنوان مثال سرطانی و نرمال) در واکنش Real time pcr نشان می دهد.
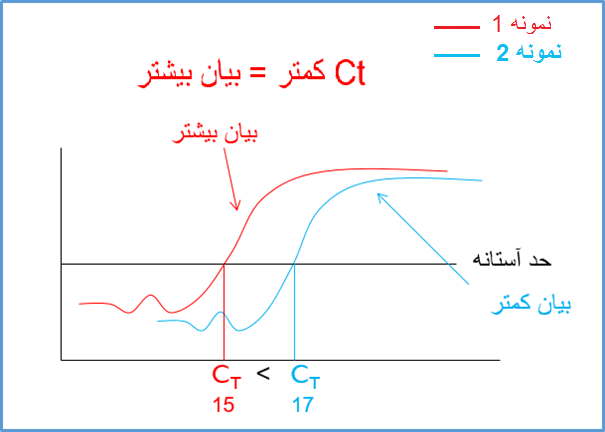
شکل 5: مفهوم سیکل آستانه در ریل تایم. در شکل فوق نمونه 1 (قرمز رنگ) در سیکل کمتر نسبت به نمونه 2 (آبی رنگ) به خط حد آستانه رسیده است. در این شکل Ct برابر نمونه 1 برابر 15 و برای نمونه 2 برابر 17 است.
آشنایی با مفهوم ژن کنترل داخلی (ژن رفرنس) در روش ریل تایم
در توضیحات قبل گفتیم که ” Ct کمتر = بیان بیشتر” و علت آن را توضیح دادیم. اما باید توجه شود که درست بودن این فرض به شرایط بسیاری بستگی دارد و رعایت نمودن همه این شروط به قدری سخت می باشند که عبارت ” Ctکمتر = بیان بیشتر” چندان قابل اعتماد نمی باشد. به عنوان مثال فرض کنید در دو نمونه شکل 5 در نمونه 1 از 4 میکرولیتر cDNA به عنوان الگو استفاده شده است در حالی که از نمونه 2 تنها یک میکرولیتر cDNA به عنوان الگو استفاده شده باشد. بنابراین میزان الگوی cDNA استفاده شده در نمونه 1 به میزان 4 برابر بیشتر از نمونه 2 بوده است و این خطا در استفاده متفاوت از میزان الگوهای cDNA بین دو نمونه مختلف سبب شده که نمونه قرمز رنگ زودتر به Ct برسد و به اشتباه میزان بیان ژن مورد بررسی در نمونه قرمز نسبت به نمونه آبی بیشتر گزارش می گردد. بنابراین با قطعیت نمی توان گفت که ” Ctکمتر = بیان بیشتر”. البته در آماده سازی واکنش ها در نمونه ها جهت Real time خطای سمپلینگ به این شدت رخ نمی دهد اما خطاهای دیگری می تواند رخ بدهد که ممکن است تفسیر اشتباه نتایج گردد. به عنوان مثال ممکن کیفیت استخراج RNA از نمونه 1 بسیار بهتر از کیفیت استخراج RNA از نمونه 2 باشد و این سبب شده که cDNA ساخته شده برای این نمونه کیفیت بیشتری داشته باشد و در نتیجه نمونه 1 زودتر به Ct برسد. و یا ممکن است کیفیت ساخت cDNA بین دو نمونه متفاوت باشد. بنابراین بروز این گونه خطاهای آزمایشگاهی سبب می شود که نتوان به Ct به عنوان یک پارامتر جهت مقایسه دو نمونه اعتماد نمود. حتی اگر فرض نمود که برای دو نمونه مختلف تمام شرایط آزمایشگاهی و کیفیت کارهای انجام شده کاملا یکسان باشد باز نمی توان شرایط یکسان را برای تمام نمونه ها در نظر گرفت برای مثال جمع آوری نمونه کافی ممکن است چندین ماه به طول انجامد که خود این سبب تغییر در میزان کیفیت الگو در واکنش Real time می گردد. برای حل این مشکل ما میتوانیم از ژن هایی استفاده کنیم که بیان یکسان آن ها در تمام نمونه ها به اثبات رسیده است (مانند ژن های GAPDH، ACTIN، و یا RPLP0). ازآنجایی که بیان بین این ژن ها در تمام نمونه ها باید یکسان باشد در صورتی که بین Ct این ژن ها در نمونه های مختلف اختلافی مشاهده گردد، به این معنی است که شرایط آزمایش برای همه نمونه ها یکسان نبوده و با توجه به Ctاین ژن ها میتوان Ct مربوط به ژن مورد بررسی را اصلاح نمود (شکل 6). در Real time به ژن های GAPDH، ACTIN، و RPLP0 ژن های کنترل داخلی (و یا ژن های رفرنس) می گویند. این ژن ها به دلیل اینکه برای حیات سلول ها ضروری می باشند معمولا دارای بیان بیشتر نسبت به سایرژن ها می باشند (Ct کمتری دارند).
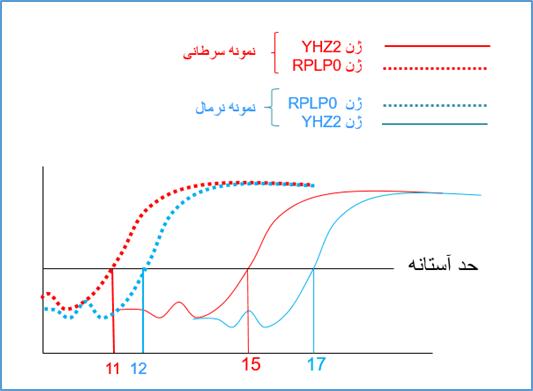
شکل 6: در شکل فوق بیان ژن YHZ2 در یک نمونه سرطانی و یک نمونه نرمال مورد بررسی قرار گرفته است همچنین ژن RPLP0 به عنوان کنترل داخلی در نظر گرفته شده است. بنابراین در ریل تایم برای هر نمونه ما بیان دو ژن را مورد برسی قرار داده ایم. در آنالیز نمونه های فوق، به اولین نکته ای که باید توجه شود بررسی کیفیت ساخت cDNA با توجه به Ct ژن کنترل داخلی در دو نمونه می باشد. در مواردی که cDNA سنتز شده دارای کیفیت مناسب می باشد، Ct ژن های کنترل داخی از 25 کمتر است. Ct بالاتر برای ژن های کنترل داخلی بیانگر کیفیت کم cDNA سنتز شده و یا شرایط بهینه نشده در ریل تایم می باشد.
دومین نکته ای که در آنالیز داده های ریل تایم باید به آن توجه نمود، Ct ژن ها در دو نمونه نرمال و سرطانی می باشد، در واکنش Real time مانند واکنش PCR معمولی باید از کنترل منفی (کنترل فاقد رشته الگو) استفاده گردد و در نتایج حاصله Ct ژن هر دو ژن ( ژن مورد بررسی و ژن رفرنس) در نمونه ها باید از Ct نمونه های کنترل منفی کمتر باشد (کنترل ها در شکل نشان داده نشده اند).
نکته سوم که در آنالیز نتایج ریل تایم به آن توجه شود این است که در واکنش Real time معمولا Ct های بالای 38 برای نمونه ها قابل قبول نیستند چون این ژن ها داری بیان بسیار بسیارکمی بوده و برای بررسی این ژن ها بهتر است کیفیت ساخت cDNA را افزایش داد تا Ctها پایین تر بیایند.
نکته:{ در یک صورت می توان Ct بالای 38 را برای ژن مورد بررسی نمونه ها مورد قبول دانست که نمونه ها دارای شرایط زیر باشند:
- Ct ژن کنترل داخلی از 21 کمتر باشد.
- از یک نمونه که ژن مورد نظر در آن بیان دارد به عنوان کنترل مثبت استفاده شود و Ct نمونه کنترل مثبت از 38 کمتر باشد.
بنابراین در صورتی که بیان یک ژن را در دو گروه بررسی می نمایید و Ct نمونه ها در گروه اول کمتر از 38 اما در گروه دوم بالاتر از 38 می باشد. در صورتی Ct گروه دوم قابل قبول می باشد که Ct ژن کنترل داخلی در نمونه های گروه دوم از 21 کمتر باشد. این نتایج نشان می دهد که ژن مورد بررسی در گروه فاقد بیان می باشد}.
آشنایی با مفهوم دلتا Ct (∆Ct) در آنالیز و تفسیر نتایج تکنیک ریل تایم (real Time PCR)
با توجه به اینکه نتایج ریل تایم ارائه شده در شکل 6 دارای همه شرایط فوق می باشد به آنالیز داده های شکل فوق بپردازیم. در این شکل همان گونه که مشاهده می شود بین Ct های ژن کنترل داخلی RPLP0 در بین نمونه های سرطانی و نرمال یک Ct تفاوت وجود دارد. بنابراین باید این تفاوت را در مقایسه Ct ژن YHZ2 بین نمونه سرطانی و کنترل اعمال نماییم و به اصطلاح داده ها (که همان Ctها هستند) را نرمال نماییم ( این نرمال نمودن به معنی نرمال یا سرطانی بودن نیست). برای نرمال کردن داده ها، Ct ژن کنترل داخلی را همه نمونه ها از Ct ژن مورد بررسی کم می کنیم. از آنجایی که داده های نرمال برای هر نمونه از کم نمودن Ct ژن کنترل داخلی از Ct ژن مورد نظر از به دست آمده است داده نرمال شده را دلتا Ct (∆Ct)می نامیم (شکل 7).
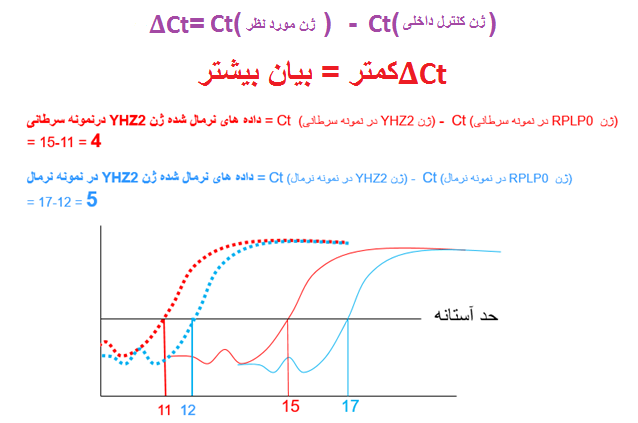
شکل 7: در این شکل محاسبات نشان می دهد که ∆Ct برای ژن YHZ2 در نمونه سرطانی برابر 4 و در نمونه نرمال برابر 5 می باشد. حال می توان گفت ∆Ct کمتر = بیان بیشتر.
آشنایی با مفهوم Fold change و نحوه محاسبه دلتا دلتا Ct (∆∆Ct) در ریل تایم
در واکنش ریل تایم هنگامی که میزان بیان یک ژن را در چند گروه مقایسه می نماییم بهترین راه بیان میزان تغییرات آن ژن در یک نمونه نسبت به نمونه دیگر fold change می باشد. مفهوم fold change همان “چند برابر” می باشد، به عبارت بهتر بیان ژن مورد نظر در نمونه های گروه اول (به عنوان مثال نمونه های سرطانی) نسبت به نمونه های گروه دوم (به عنوان مثال نمونه های نرمال) چند برابر افزایش و یا کاهش یافته است؟ این جمله به این مفهوم است که اگر بیان ژن مورد بررسی را در نمونه نرمال برابر 1 در نظر بگیریم، بیان آن در نمونه سرطانی برابر چه میزانی خواهد بود؟
قبل از توضیح محاسبه fold change در real time به این مثال توجه فرمایید وزن فرد A برابر 60 کیلوگرم و وزن فرد B برابر120 گیلوگرم است، پس Fold change وزن فرد B نسبت فرد A برابر است با 2 برابر (120 تقسیم بر 60). توجه داشته باشید که در اعلام نتایج باید Fold change فرد A نسبت خودش هم ذکر شود زیرا برای بررسی های آماری مورد نیاز می باشد (شکل 8).
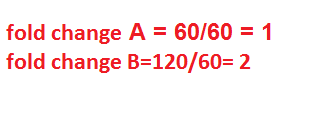
شکل 8: نحوه محاسبه fold change برای وزن.
باید توجه داشت که در مثال فوق، وزن ارائه شده برای افراد در مقیاس خطی بوده (لگاریتمی نبوده) بنابراین داده ها بر هم تقسیم شده اند (شکل 8). اما داده های ∆Ct از نوع لگاریتمی برپایه 2 می باشد ( زیرا در real time هر سیکل محصول ما 2 برابر می شود). بنابراین برای محاسبه fold change در ریل تایم باید ∆Ct ها را از هم تفریق نمود. پارامتر جدید را که از تفریق ∆Ct ژن مورد نظر در نمونه سرطانی از ∆Ct نمونه غیر سرطانی به دست آمده است را دلتا دلتا Ct (∆∆Ct)می نامیم (شکل 9).

شکل 9: نحوه محاسبه دلتا دلتا Ct (∆∆Ct) در ریل تایم. در شکل 9 میزان ∆∆Ct برای نمونه سرطانی برابر با 1- و برای نمونه غیرسرطانی برابر با 0 می باشد. بنابراین بیان ژن مورد بررسی در نمونه سرطانی نسبت به نمونه غیر سرطانی بیشتر است. به عبارتی می توان گفت که ” ∆∆Ct کمتر = بیان بیشتر”.
در توضیحات قبلی ابتدا ذکر نمودیم که ” Ct کمتر = بیان بیشتر” و سپس آن را اصلاح نمودیم و ذکر کردیم که “∆Ct کمتر = بیان بیشتر” و سپس برای محاسبه fold change میزان ∆∆Ct را محاسبه نمودیم و ذکر نمودیم که ” ∆∆Ct کمتر = بیان بیشتر”، اما در ادامه می خواهیم بگوییم ” fold change بیشتر = بیان بیشتر” . برای این منظور ابتدا باید مقدار ∆∆Ct را در عدد (1-) ضرب نمود پس اکنون می گوییم ” -∆∆Ct بیشتر= بیان بیشتر”.
همان گونه که قبلا گفته شد داده های Ct، ∆Ct ، ∆∆Ct و -∆∆Ct بر اساس لگاریتم بر پایه 2 هستند بنابراین -∆∆Ct همان fold change به صورت لگاریتمی بر پایه2 می باشد. حال برای اینکه -∆∆Ct را به حالت خطی در اورد، باید 2 را توان -∆∆Ct رساند. مقادیر حاصله بیانگر fold change برای هر نمونه خواهد بود (شکل 10).

شکل 10: نحوه محاسبه fold chane در داده های ریل تایم. در نهایت Fold change برای نمونه نرمال برابر 1 و برای نمونه سرطانی برابر 2 می باشد یعنی بیان ژن در نمونه سرطانی 2 برابر نمونه نرمال می باشد.
نکته: همیشه fold change برای گروه کنترل برابر 1 خواهد شد.
آشنایی با فرمول های مربوطه به محاسبه فولد چنج و آنالیز ریل تایم
فرمول های فوق را می توان به صورت خلاصه در شکل 11 نشان داد.

شکل 11: فرمول محاسبه fold change برای نتایج ریل تایم.
آموزش آنالیز داده های ریل تایم (Real time) و تفسیر آن ها به صورت عملی
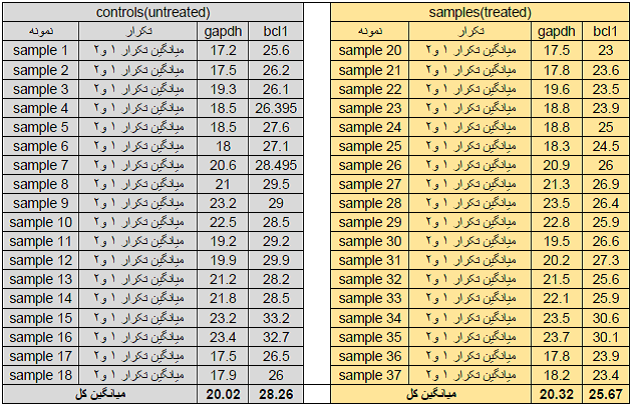
حال که با مفاهیم اولیه آنالیز داده ها در تکنیک Real time آشنا شدید، یک نمونه از داده های Real time را مورد آنالیز قرار می دهیم. در این بررسی بیان ژن bcl1 نمونه های سرطانی نسبت به نمونه نرمال مورد بررسی قرار گرفته است. از آن جاییکه یکی از مسائل مهم در آزمایش های بیولوژیکی اصل تکرار آزمایش می باشد بنابر این هر نمونه را دو بار ( در یک واکنش Real time) بررسی قرار گرفته است. جدول Ct ها برای نمونه ها و تکرارها در جدول زیر ارائه شده است.
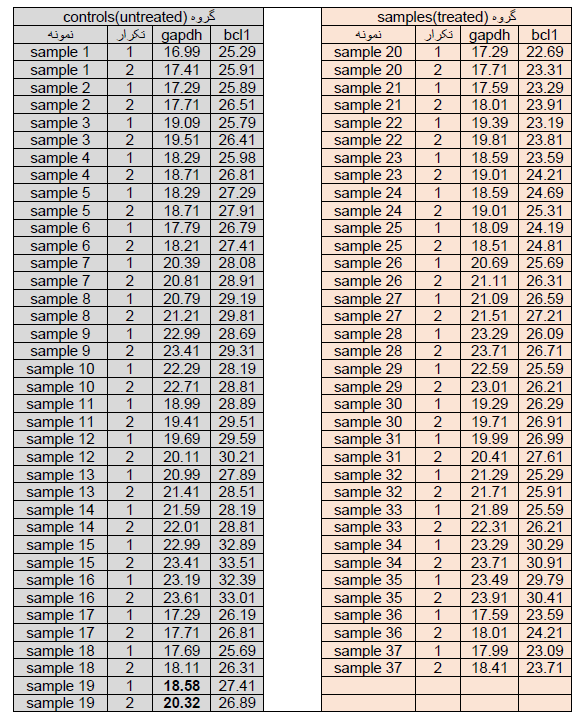
جدول 1.
آموزش نحوه بررسی کیفیت نتایج ریل تایم
قبل از آنالیز داده ها در ابتدا باید کیفیت نمونه ها در چند مرحله چک شوند.
مرحله اول: در بیشتر نمونه ها Ct ژن bcl1 از 38 کمتر می باشد. توجه کنید که در بیشتر نمونه های هر دو گروه نرمال وسرطانی نباید Ct ژن bcl1 از 38 بیشتر باشد. چون Ct بالای 38 درReal time قابل قبول نمی باشد. اما اگر در گروه نرمال همه Ctها بالای 38 بود اما در گروه سرطانی اکثرا زیر 38 باشد این Ct ها قابل قبول هستند زیرا می توان گفت این ژن در گروه نرمال بیان ندارد اما در گروه تومری بیان دارد ( یا برعکس).
مرحله دوم: برای هر دو ژن bcl1 و gapdh در هر دو تکرار برای یک نمونه، نباید اختلاف Ct بیش از 1 باشد. در جدول 1 اختلاف Ct ژن gapdh در تکرارهای نمونه شماره 19 برابر 1.74 می باشد بنابراین این نمونه باید از آنالیز ها بیرون گذاشته شود (به شکل زیر توجه کنید).
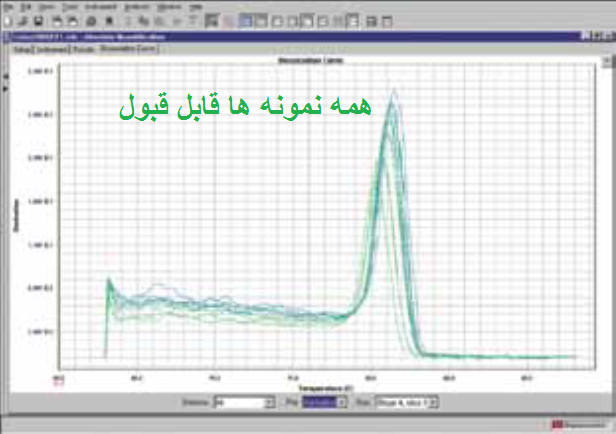
شکل فوق: در این شکل 10 نمونه بررسی شده اند که با رنگ های مختلف نشان داده شده اند. هر نمونه 5 بار تکرار شده است و مشاهده می شود که منحنی همه تکرارها برای هر نمونه برروی هم منطبق می باشند. اما در صورتی که یک تکرار منحنی آن با تکرار های دیگر منطبق نباشد باید آن تکرار از بررسی ها حذف شود.
آموزش تفسیر منحنی ذوب (melt curve) در ریل تایم پی سی ار
مرحله سوم: بررسی منحنی ذوب (melt curve) نمونه ها. در صورتی که از رنگ های فلورسنت مانند سایبرگرین در ریل تایم استفاده نموده اید می توانید منحنی ذوب را برای هر نمونه مورد بررسی قرار دهید. از آن جاییکه هر ژن دارای منحنی ذوب خاص خود می باشد بنابراین منحنی های یک ژن در تمام نمونه ها باید با هم منطبق باشند و همچنین باید تمام منحنی ها تک قله باشند. به شکل های زیر توجه کنید.
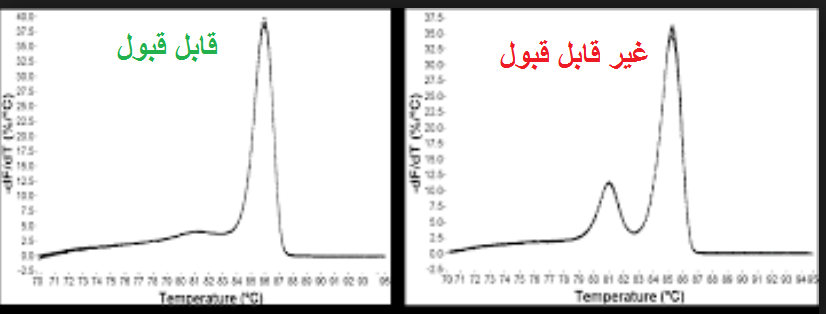
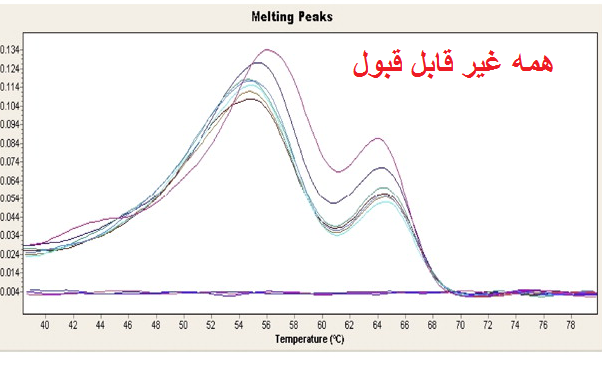
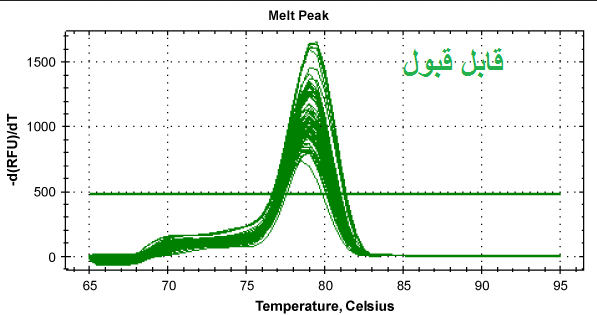
شکل 12: نمونه هایی از منحنی ذوب قابل قبول و غیر قابل قبول. توجه کنید که همه قله های یک ژن باید روی یک دما باشند ولی نیازی نیست ارتفاع قله ها یکسان باشد.
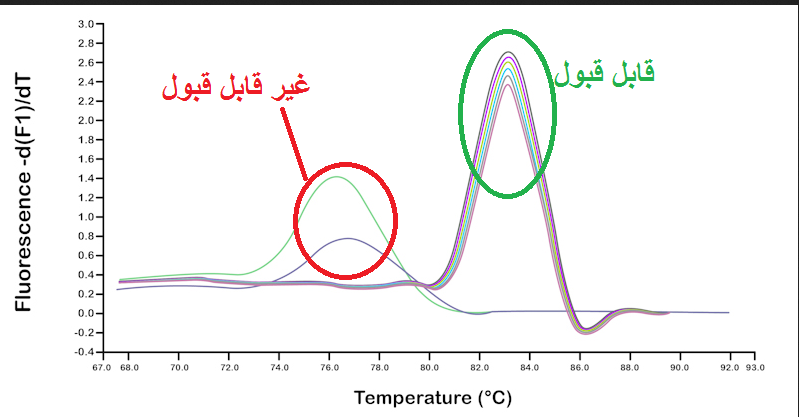
شکل 13: در آنالیز داده های ریل تایم نمونه هایی که منحنی ذوب آن ها برای هر دو ژن با سایر نمونه ها منطبق نیست باید از بررسی حذف شوند. توجه کنید که منحنی ذوب ژن bcl1 و ژن کنترل داخلی باید جدا گانه مورد بررسی قرار گیرند و نیازی نیست منحنی های این دو ژن با هم منطبق باشد.
بعد از حذف نمونه های غیرقابل قبول نوبت به آنالیز داده ها می رسد در این مرحله چون تمام تکرارها در یک واکنش Real time به صورت هم زمان بررسی شده اند باید از Ct های حاصله برای هر نمونه میانگین گرفت و با ct میانگین کار کرد.
نکته: در مرحله میانگین گرفتن توجه شود که، در این مثال در یک واکنش ریل تایم، هر نمونه 2 بار همزمان تکرار شده است بنابراین باید برای هر نمونه میانگین گرفت. اما اگر نمونه ها چند نوع باشند به عنوان مثال یک تیمار که دو بارتکرارشده است، دیگر نباید بین تکرار تیمارها میانگین گرفت.
نکته: در صورتی که یک ریل تایمی که قبلا اجرا شده دو باره تکرار شود. نباید بین Ct نتایج قدیم و جدید میانگین گرفته شود. نحوه آنالیز این داده ها در ادامه توضیح داده خواهد شد.
جدول 2: در جدول فوق برای همه نمونه ها، میانگین گرفته شده است.
اکنون مرحله بعد محاسبه Fold change می باشد. براساس جدول 2 و فرمول 1 می توان Fold change ژن bcl1 را بین دو گروه محاسبه نمود (شکل 14).

شکل 14: میزان fold change محاسبه شده بر اساس فرمول 1 برابر است با 7.46. بنابراین ژن bcl1 در نمونه توموری نسبت به نمونه نرمال 7.46 برابر افزایش بیان داشته است.
تفاوت Ct با Cp در آنالیز داده های ریل تایم چیست؟
همانگونه که قبلا ذکر نمودیم Ct به این مفهوم است که در چه سیکلی محصولات real time از یک حد آستانه (threshold) فراتر می رود. در بخی از نرم افزارهای ریل تایم از Cp به جای Ct استفاده می نمایند. Cp مخفف crossing point می باشد و به مفهوم سیکلی می باشد که در آن میزان محصولات real time از یک نقطه خاص (crossing point) فراتر می رود. در عمل تفاوتی بین آنالیز داده ها براساس Ct و یا Cp وجود ندارد. فرمول 2 شکل اصلاح شده فرمول 1 بر اساس Cp می باشد.

فرمول 2: فرمول اصلاح شده محاسبه fold change بر اساس Cp.
نکته: با کمی تغییر در فرمول 1 و یا فرمول 2 ، فرمول 3 جهت محاسبه fold change به دست می آید. البته میزان fold change محاسبه با هر سه فرمول یکسان خواهد بود.

فرمول 3.
در شکل 15 میزان fold change برای داده های جدول 2 براساس فرمول 3 محاسبه گردیده است.

شکل 15: محاسبه میزان fold change برای داده های جدول 2 براساس فرمول 3. همانگونه که قابل مشاهده می باشد fold change محاسبه شده در این فرمول با فرمول های قبلی تفاوتی ندارد.
ضرورت محاسبه میزان کارایی PCR و پرایمرها برای آنالیز نتایج ریل تایم.
در فرمول های 1، 2 و 3 از عدد 2 در فرمول استفاده شده است چرا؟ به دلیل اینکه در واکنش ریل تایم (همانند واکنش PCR) میزان محصولات در هر سیکل باید 2 برابر شود.
استفاده از فرمولهای 1 تا 3 برای محاسبه fold change در شرایطی صحیح است که میزان کارایی همه پرایمرها برابر با 100درصد باشد. 100درصد بودن کارایی پرایمرها به این معنی است که محصولات ریل تایم در هر سیکل از واکنش، 2 برابر می شوند، اما همیشه این گونه نیست و در بسیاری از موارد کارایی پرایمرها از 100درصد کمتر است بنابراین نمی توان از عدد 2 در فرمول 1 استفاده نمود. به عنوان مثال اگر کارایی پرایمرهای ژن هدف و ژن کنترل داخلی برابر با 80درصد باشد بنابراین در فرمول 1 به جای عدد 2 باید از عدد 1.8 (1+0.8) استفاده نمود.
فرمول 4 شکل اصلاح شده فرمول 3 می باشد که در آن میزان کارایی PCR در فرمول لحاظ شده است.
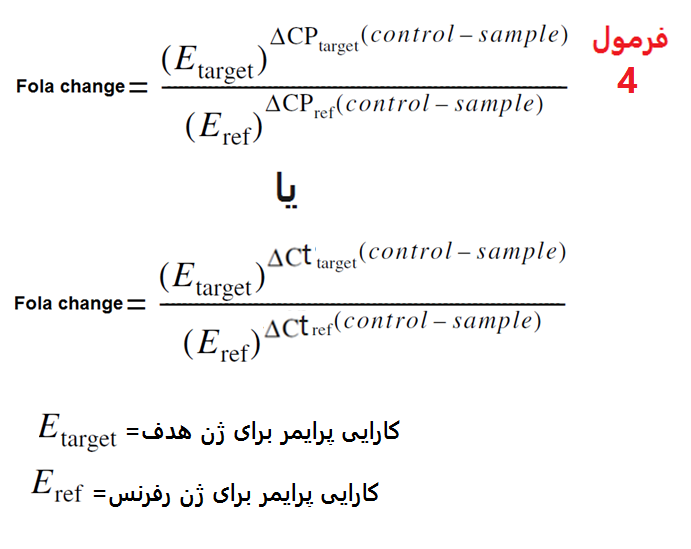
فرمول 4: فرمول اصلاح شده محاسبه fold change در ریل تایم.
در فرمول شماره 4 مقدار E جایگزین عدد 2 (در فرمول 3( شده است که E برابر با کارایی PCR (PCR Efficiency) برای ژن های هدف و رفرنس می باشد. E برابر است با 1 به علاوه میزان کارایی پرایمرها (فرمول 5).

فرمول 5: نحوه محاسبه E.
در فرمول 5 کارایی پرایمرها بین 100درصد (عدد 1) تا صفر درصد (عدد 0) متغیر می باشد. بنابراین میزان E بین 2 (در بهترین حالت تکثیر محصولات ) تا 1 ( عدم تکثیر محصولات) متغیر می باشد. پس در فرمول 3 اگر E برابر با 1 باشد، پرایمرها کارایی نداشته و محصولی تکثیر نشده است.
در صورتی که میزان کارایی پرایمر برای ژن مورد بررسی برابر با 0.930 و میزان کارایی پرایمر برای ژن رفرنس برابر با 0.866 باشد میزان fold change محاسبه شده برای داده های جدول 2 و با استفاده از فرمول 4 برابر با 6.67 خواهد بود (شکل 16)

شکل 16: میزان fold change محاسبه شده برای داده های جدول 2 و فرمول 4. همانگونه که مشاهده می نمایید میزان fold change محاسبه شده بدون در نظر گرفتن کارایی پرایمرها (فرمول 3) برابر 7.46 محاسبه شده بود اما با به کارگیری کارایی پرایمرها (فرمول 4) fold change به صورت دقیق تر و برابر 6.67 محاسبه شده است. در مجموع می توان گفت ضرورت محاسبه میزان کارایی پرایمرها و یا میزان E در محاسبه دقیق میزان fold change برای نتایج ریل تایم می باشد.
برای محاسبه میزان کارایی پرایمرها و یا میزان E از دو روش می توان استفاده نمود.
- استفاده از رقت های متوالی در واکنش ریل تایم و ترسیم منحنی استاندارد و محاسبه شیب خط
- استفاده از نرم افزار linregPCR
روش اول نیازمند تهیه رقت های متوالی از نمونه ها می باشد بنابراین روش اول نیازمند صرف هزینه های بیشتر می باشد اما در روش دوم با استفاده از نرم افزار linregPCR می توان نتایج ریل تایم حاصل از نمونه های اصلی را مورد بررسی قرار داد و به این طریق میزان کارایی پرایمرها و میزان E را محاسبه نمود.
آشنایی با نرم افزار LinRegPCR
نرم افزار LinRegPCR یک نرم افزار برای بررسی داده های ریل تایم به منظور محاسبه کارایی پرایمرها و PCR ( E یا PCR Efficiency) می باشد. این نرم افزار داده های ریل تایم را بررسی نموده و بوسیله رگرسیون خطی و تخمین شیب (slope) خط رگرسیون برای هر نمونه، کارایی پرایمرها و PCR ( E یا PCR Efficiency) را محاسبه می نماید.